EnrichMap draws an enrichment map – a gene-set similarity network
where each node is an enriched term, node size encodes the number of
associated genes, node fill colour encodes cluster membership (detected
via igraph community detection), and edge thickness encodes the number
of overlapping genes between term pairs. The plot uses a force-directed
layout to arrange terms, and ggforce hull annotations group terms into
clusters. Keyword or term-description labels appear in the legend.
EnrichNetwork draws an enrichment network – a term-gene bipartite
graph where term nodes are shown as numbered circles and gene nodes as
labelled rectangles. Gene node colours are blended from the colours of
all terms they belong to. A force-directed layout positions the nodes,
with optional overlap adjustment for better readability.
Both functions accept enrichment results from clusterProfiler or Enrichr
(the latter is auto-detected and preprocessed via
prepare_enrichr_result()).
Usage
EnrichMap(
data,
in_form = c("auto", "clusterProfiler", "clusterprofiler", "enrichr"),
split_by = NULL,
split_by_sep = "_",
top_term = 10,
metric = "p.adjust",
layout = "fr",
minchar = 2,
cluster = "fast_greedy",
show_keyword = FALSE,
nlabel = 4,
character_width = 50,
mark = "ellipse",
label = c("term", "feature"),
labelsize = 5,
expand = c(0.4, 0.4),
theme = "theme_this",
theme_args = list(),
palette = "Paired",
palcolor = NULL,
palreverse = FALSE,
alpha = 1,
aspect.ratio = 1,
legend.position = "right",
legend.direction = "vertical",
title = NULL,
subtitle = NULL,
xlab = NULL,
ylab = NULL,
seed = 8525,
combine = TRUE,
nrow = NULL,
ncol = NULL,
byrow = TRUE,
axes = NULL,
axis_titles = axes,
guides = NULL,
design = NULL,
...
)
EnrichNetwork(
data,
in_form = c("auto", "clusterProfiler", "clusterprofiler", "enrichr"),
split_by = NULL,
split_by_sep = "_",
top_term = 10,
metric = "p.adjust",
character_width = 50,
layout = "fr",
layoutadjust = TRUE,
adjscale = 60,
adjiter = 100,
blendmode = "blend",
labelsize = 5,
theme = "theme_this",
theme_args = list(),
palette = "Paired",
palcolor = NULL,
palreverse = FALSE,
alpha = 1,
aspect.ratio = 1,
legend.position = "right",
legend.direction = "vertical",
title = NULL,
subtitle = NULL,
xlab = NULL,
ylab = NULL,
seed = 8525,
combine = TRUE,
nrow = NULL,
ncol = NULL,
byrow = TRUE,
axes = NULL,
axis_titles = axes,
guides = NULL,
design = NULL,
...
)Arguments
- data
A data frame containing enrichment results in clusterProfiler format (see
EnrichMapfor the expected columns). If you have enrichment results from multiple databases, you can combine them into one data frame and add a column (e.g.Database) to indicate the source. Usesplit_by = "Database"to plot them side by side.- in_form
A character string specifying the input format. When
"auto"(default), the function infers the format from the column names. Other options are"clusterProfiler","clusterprofiler", and"enrichr".- split_by
The column(s) to split data by and plot separately.
- split_by_sep
The separator for multiple split_by columns. See
split_by- top_term
An integer specifying the maximum number of terms to include. Terms are ranked by
metric(ascending). Default100.- metric
A character string specifying the significance metric used for top-term selection and node scoring:
"p.adjust"(default) or"pvalue". The value is transformed as \(-\log_{10}(metric)\).- layout
A character string naming the igraph layout algorithm. Built-in shortcuts:
"circle","tree","grid". Otherwise, the suffix passed tolayout_with_<layout>in igraph (e.g."fr"for Fruchterman-Reingold,"kk"for Kamada-Kawai). Default"fr".- minchar
An integer specifying the minimum character length for words to be included as keywords when
show_keyword = TRUE. Default2.- cluster
A character string naming the igraph community detection algorithm. The suffix passed to
cluster_<cluster>in igraph (e.g."fast_greedy","walktrap","edge_betweenness","infomap"). Default"fast_greedy".- show_keyword
A logical value. When
TRUE, theDescriptiontext is tokenized and the most significant words per cluster are shown as keywords. WhenFALSE(default), the original term descriptions are used as labels.- nlabel
An integer specifying the number of keywords or term descriptions to show per cluster in the legend labels. Default
4.- character_width
An integer specifying the maximum width (in characters) at which keyword labels are wrapped via
strwrap(width = character_width). Default50.- mark
A character string naming the ggforce hull function. One of
"ellipse"(default),"rect","circle", or"text"– passed as the suffix togeom_mark_<mark>.- label
A character string specifying what information to display in the legend labels. Either
"term"(default; shows top term descriptions/keywords per cluster) or"feature"(shows top gene symbols per cluster).- labelsize
A numeric value specifying the font size of the cluster labels drawn by the ggforce mark layer. Default
5.- expand
The values to expand the x and y axes. It is like CSS padding. When a single value is provided, it is used for both axes on both sides. When two values are provided, the first value is used for the top/bottom side and the second value is used for the left/right side. When three values are provided, the first value is used for the top side, the second value is used for the left/right side, and the third value is used for the bottom side. When four values are provided, the values are used for the top, right, bottom, and left sides, respectively. You can also use a named vector to specify the values for each side. When the axis is discrete, the values will be applied as 'add' to the 'expansion' function. When the axis is continuous, the values will be applied as 'mult' to the 'expansion' function. See also https://ggplot2.tidyverse.org/reference/expansion.html
- theme
A character string or a theme class (i.e. ggplot2::theme_classic) specifying the theme to use. Default is "theme_this".
- theme_args
A list of arguments to pass to the theme function.
- palette
A character string specifying the palette to use. A named list or vector can be used to specify the palettes for different
split_byvalues.- palcolor
A character string specifying the color to use in the palette. A named list can be used to specify the colors for different
split_byvalues. If some values are missing, the values from the palette will be used (palcolor will be NULL for those values).- palreverse
A logical value indicating whether to reverse the palette. Default is FALSE.
- alpha
A numeric value specifying the transparency of the plot.
- aspect.ratio
A numeric value specifying the aspect ratio of the plot.
- legend.position
A character string specifying the position of the legend. if
waiver(), for single groups, the legend will be "none", otherwise "right".- legend.direction
A character string specifying the direction of the legend.
- title
A character string specifying the title of the plot. A function can be used to generate the title based on the default title. This is useful when split_by is used and the title needs to be dynamic.
- subtitle
A character string specifying the subtitle of the plot.
- xlab
A character string specifying the x-axis label.
- ylab
A character string specifying the y-axis label.
- seed
The random seed to use. Default is 8525.
- combine
Whether to combine the plots into one when facet is FALSE. Default is TRUE.
- nrow
A numeric value specifying the number of rows in the facet.
- ncol
A numeric value specifying the number of columns in the facet.
- byrow
A logical value indicating whether to fill the plots by row.
- axes
A string specifying how axes should be treated. Passed to
patchwork::wrap_plots(). Only relevant whensplit_byis used andcombineis TRUE. Options are:'keep' will retain all axes in individual plots.
'collect' will remove duplicated axes when placed in the same run of rows or columns of the layout.
'collect_x' and 'collect_y' will remove duplicated x-axes in the columns or duplicated y-axes in the rows respectively.
- axis_titles
A string specifying how axis titltes should be treated. Passed to
patchwork::wrap_plots(). Only relevant whensplit_byis used andcombineis TRUE. Options are:'keep' will retain all axis titles in individual plots.
'collect' will remove duplicated titles in one direction and merge titles in the opposite direction.
'collect_x' and 'collect_y' control this for x-axis titles and y-axis titles respectively.
- guides
A string specifying how guides should be treated in the layout. Passed to
patchwork::wrap_plots(). Only relevant whensplit_byis used andcombineis TRUE. Options are:'collect' will collect guides below to the given nesting level, removing duplicates.
'keep' will stop collection at this level and let guides be placed alongside their plot.
'auto' will allow guides to be collected if a upper level tries, but place them alongside the plot if not.
- design
Specification of the location of areas in the layout, passed to
patchwork::wrap_plots(). Only relevant whensplit_byis used andcombineis TRUE. When specified,nrow,ncol, andbyroware ignored. Seepatchwork::wrap_plots()for more details.- ...
Additional arguments.
- layoutadjust
A logical value. When
TRUE(default), appliesadjust_network_layout()after the initial layout to reduce node overlap based on label width and a repulsion simulation.- adjscale
A numeric value controlling the scale of the layout adjustment. Passed as the
scaleargument toadjust_network_layout(). Default60.- adjiter
A numeric value controlling the number of iterations for the layout adjustment. Passed as the
iterargument toadjust_network_layout(). Default100.- blendmode
A character string specifying how gene colours are computed from the colours of the terms they belong to. One of
"blend"(default),"average","multiply", or"screen". Passed toblend_colors().
Value
A ggplot object (single plot), a patchwork /
wrap_plots object (when split_by is provided and
combine = TRUE), or a list of ggplot objects (when
split_by is provided and combine = FALSE).
A ggplot object (single plot), a patchwork /
wrap_plots object (when split_by is provided and
combine = TRUE), or a list of ggplot objects (when
split_by is provided and combine = FALSE).
split_by Workflow (EnrichMap)
When split_by is provided, EnrichMap() executes the
following pipeline:
Argument validation –
validate_common_args()checks the seed.Input format detection –
match.arg()resolvesin_form;"auto"mode infers the format from column names.Enrichr preprocessing – when format is
"enrichr", callsprepare_enrichr_result()to rename columns and infer GeneRatio/BgRatio.Split column resolution –
check_columns()validatessplit_by(force_factor, allow_multi, concat_multi).Data splitting – splits
databysplit_bylevels, preserving factor level order.Per-split palette/colour –
check_palette()andcheck_palcolor()resolve per-split palette and colour overrides.Per-split legend –
check_legend()resolveslegend.positionandlegend.directionper split.Per-split title – when
titleis a function, it receives the default title (the split level name); otherwisetitle %||% split_levelis used.Dispatch – each split subset is passed to
EnrichMapAtomicwith its resolved parameters.Combination –
combine_plots()assembles the list of plots viapatchwork::wrap_plots, honouringnrow/ncol/byrow/axes/axis_titles/guides/design.
split_by Workflow (EnrichNetwork)
When split_by is provided, EnrichNetwork() executes the
same pipeline as EnrichMap() above, but dispatches each split
subset to EnrichNetworkAtomic.
Examples
# \donttest{
data(enrich_example)
EnrichMap(enrich_example)
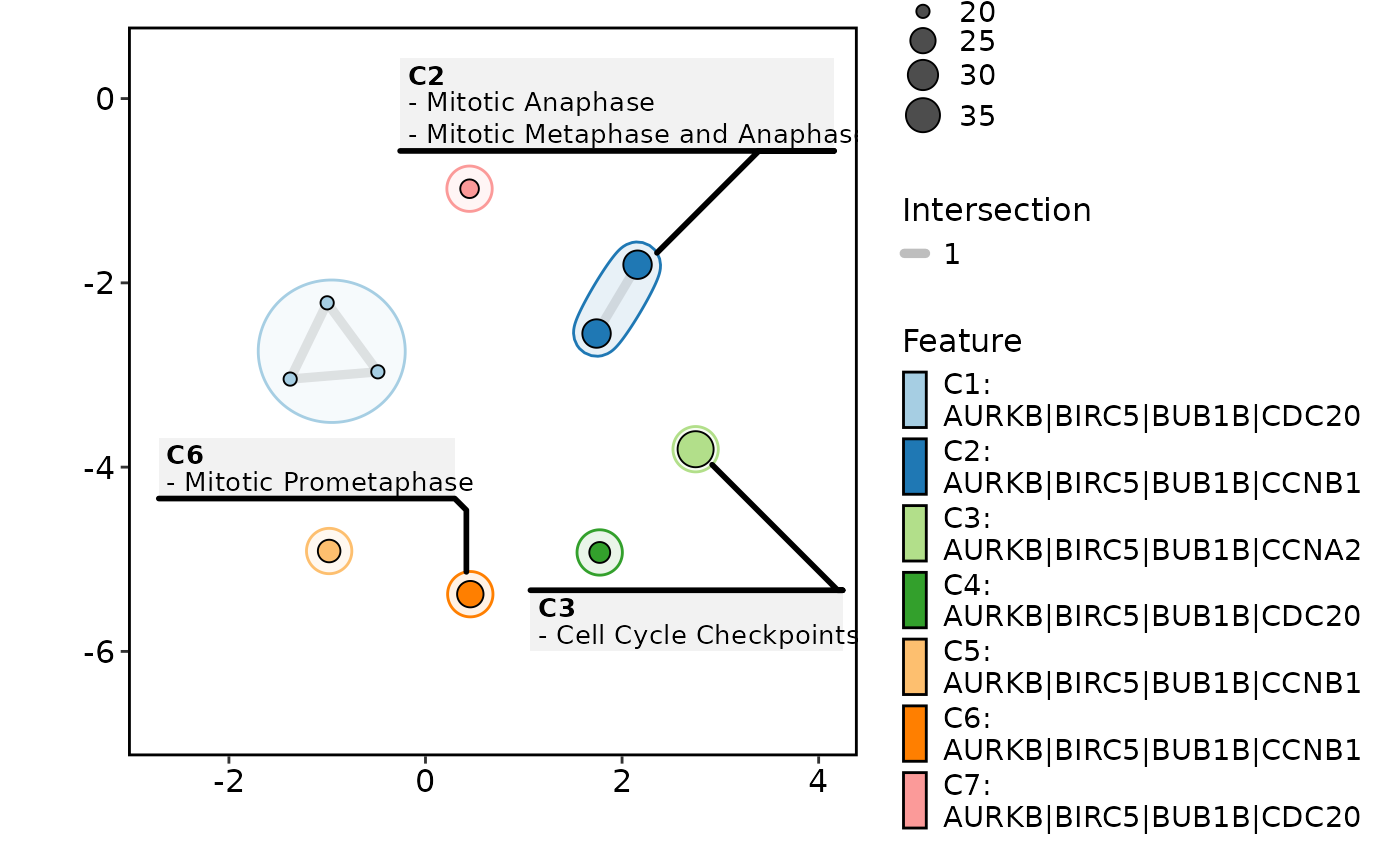 EnrichMap(enrich_example, label = "feature")
EnrichMap(enrich_example, label = "feature")
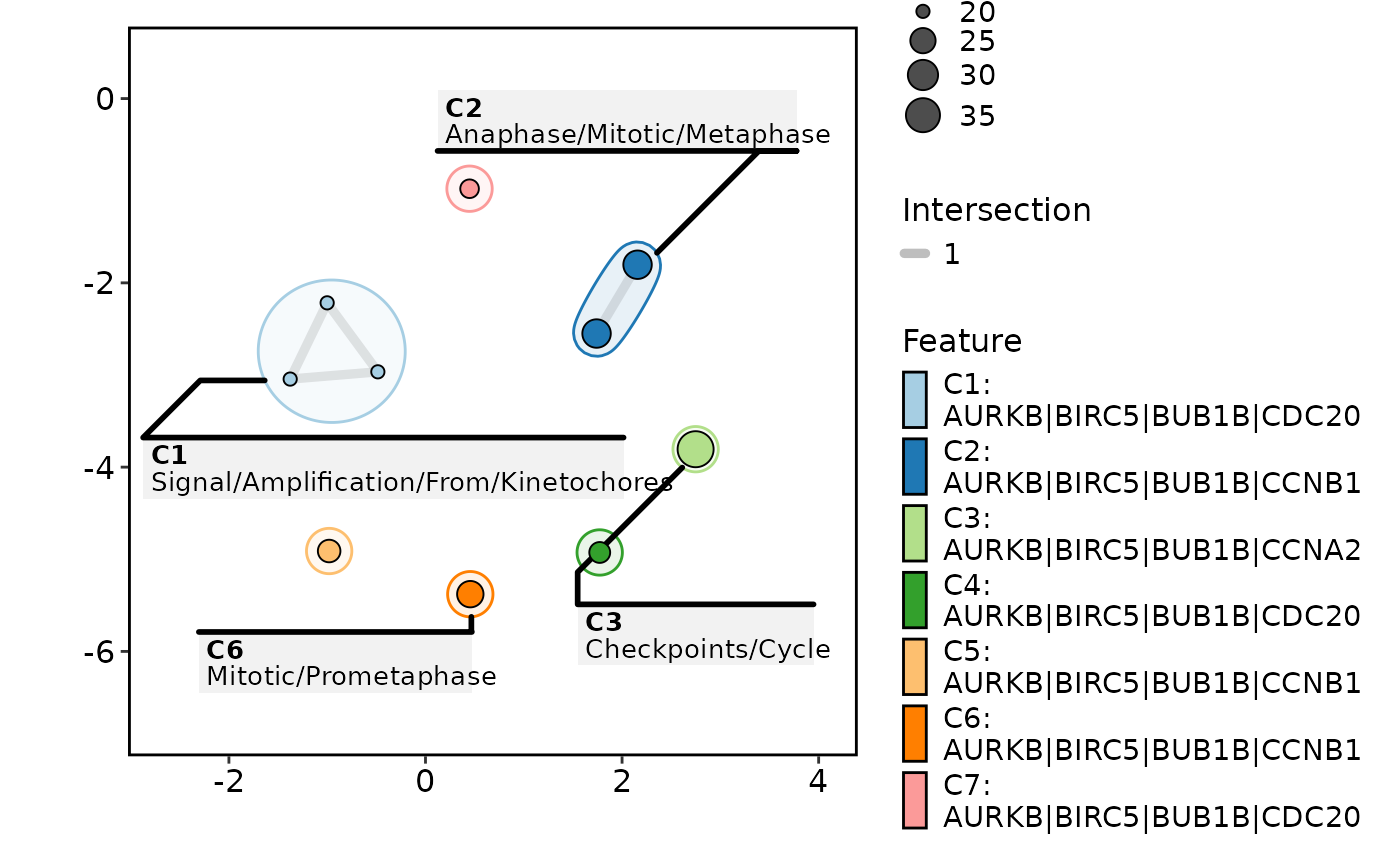
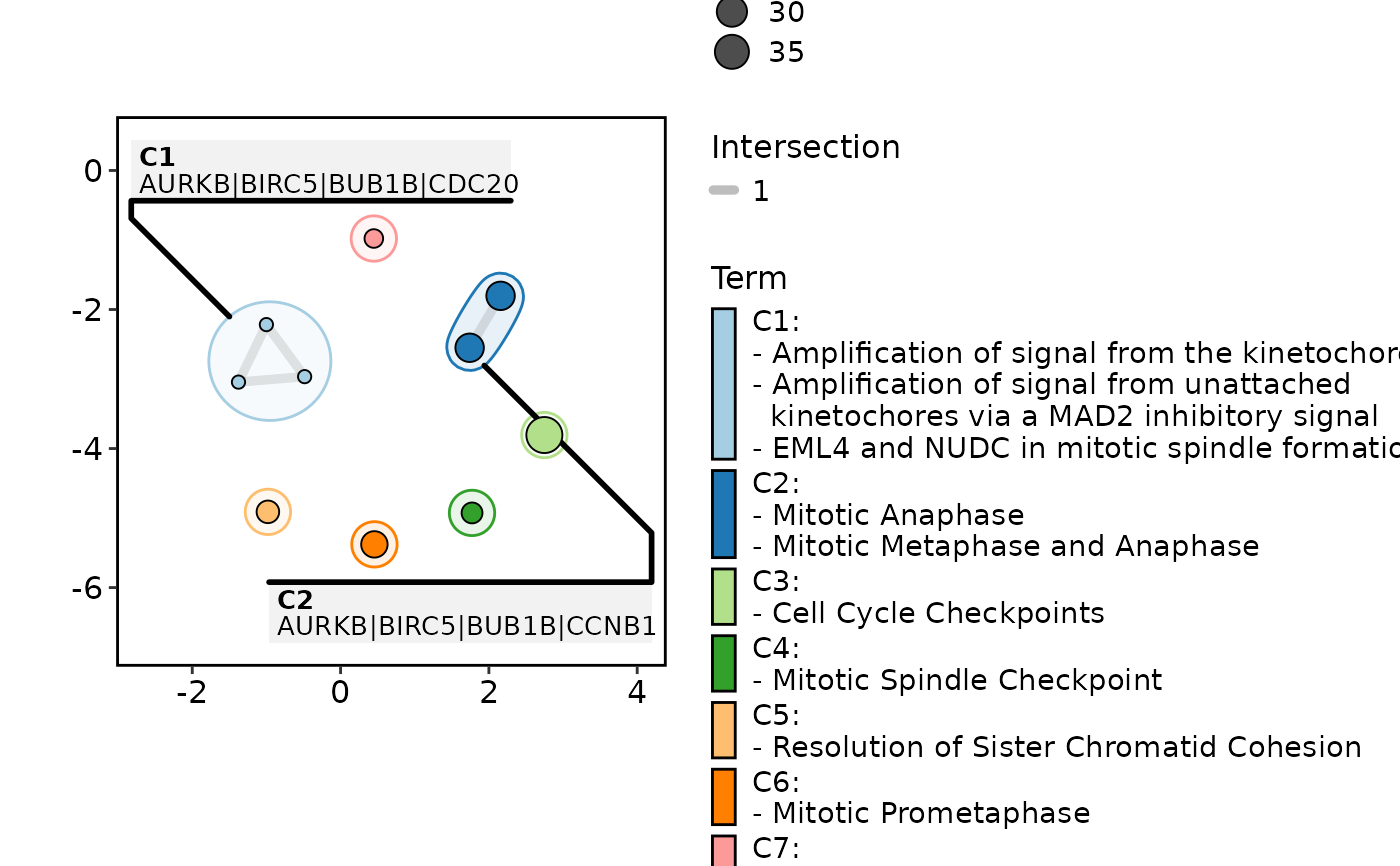 EnrichMap(enrich_example, show_keyword = TRUE, label = "term")
EnrichMap(enrich_example, show_keyword = TRUE, label = "term")
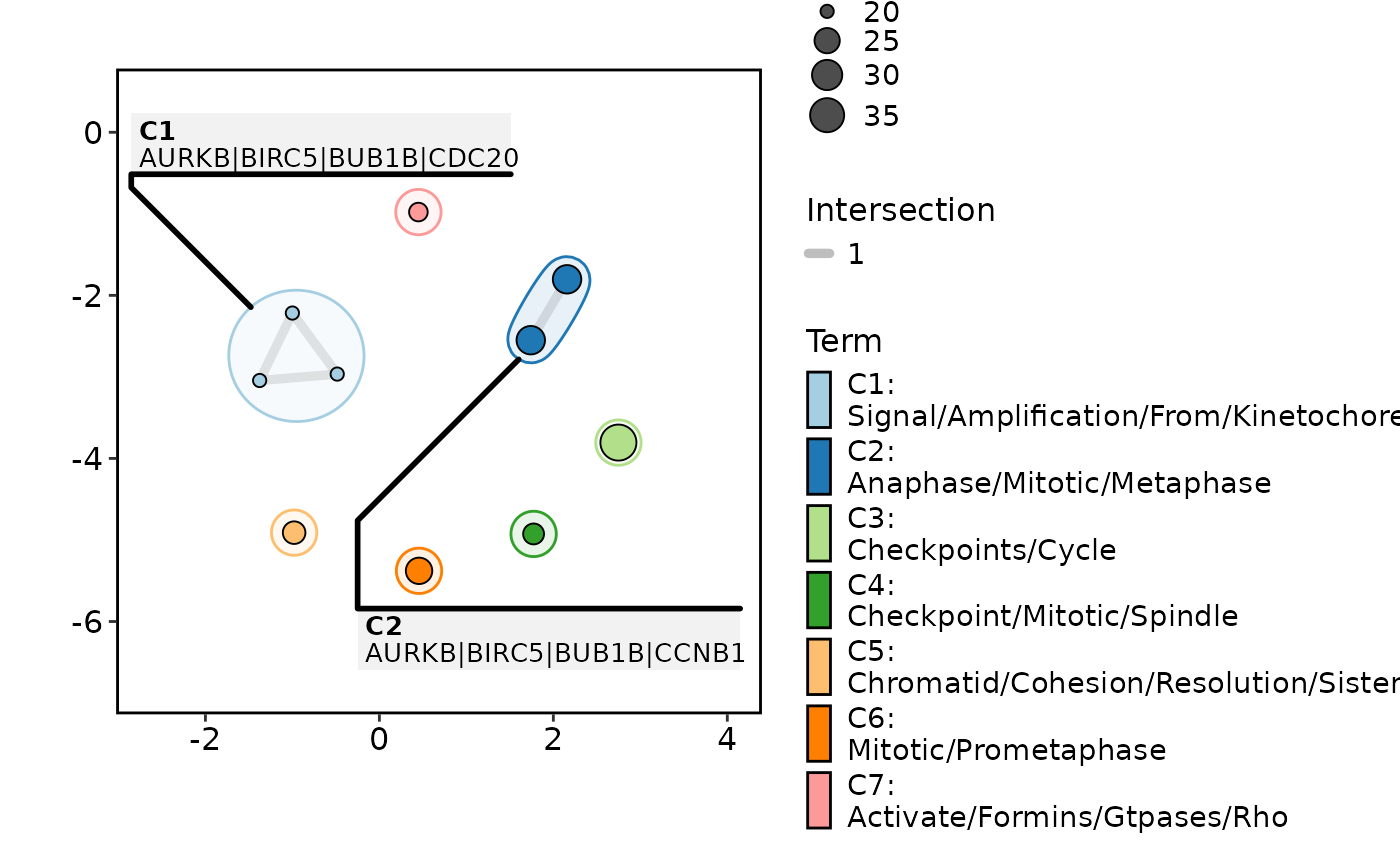
 EnrichMap(enrich_example, show_keyword = TRUE, label = "feature")
EnrichMap(enrich_example, show_keyword = TRUE, label = "feature")
 data(enrich_multidb_example)
EnrichMap(enrich_multidb_example, split_by = "Database")
data(enrich_multidb_example)
EnrichMap(enrich_multidb_example, split_by = "Database")
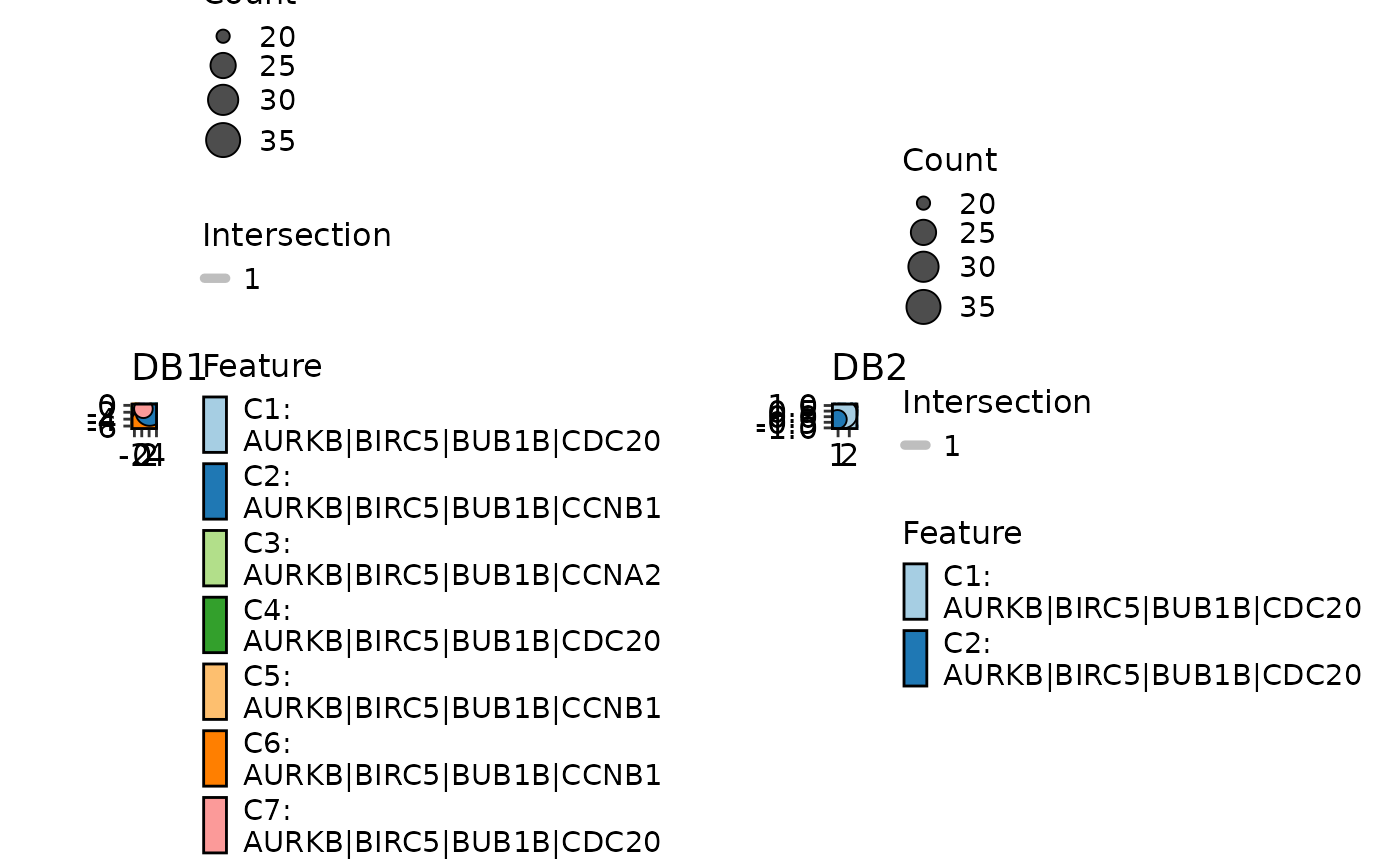 EnrichMap(enrich_multidb_example, split_by = "Database",
palette = list(DB1 = "Paired", DB2 = "Set1"))
EnrichMap(enrich_multidb_example, split_by = "Database",
palette = list(DB1 = "Paired", DB2 = "Set1"))
 # }
# \donttest{
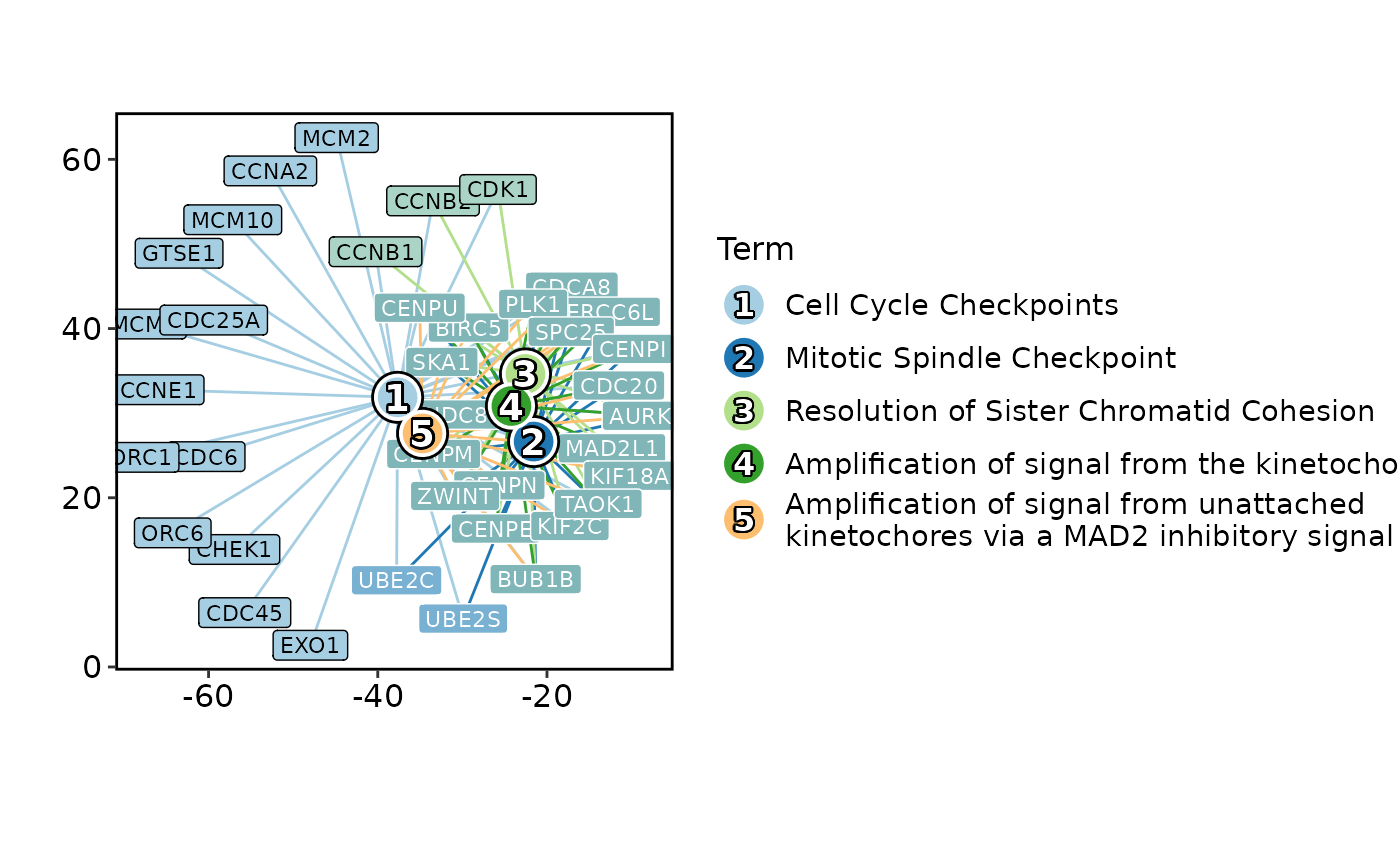
EnrichNetwork(enrich_example, top_term = 5)
# }
# \donttest{
EnrichNetwork(enrich_example, top_term = 5)
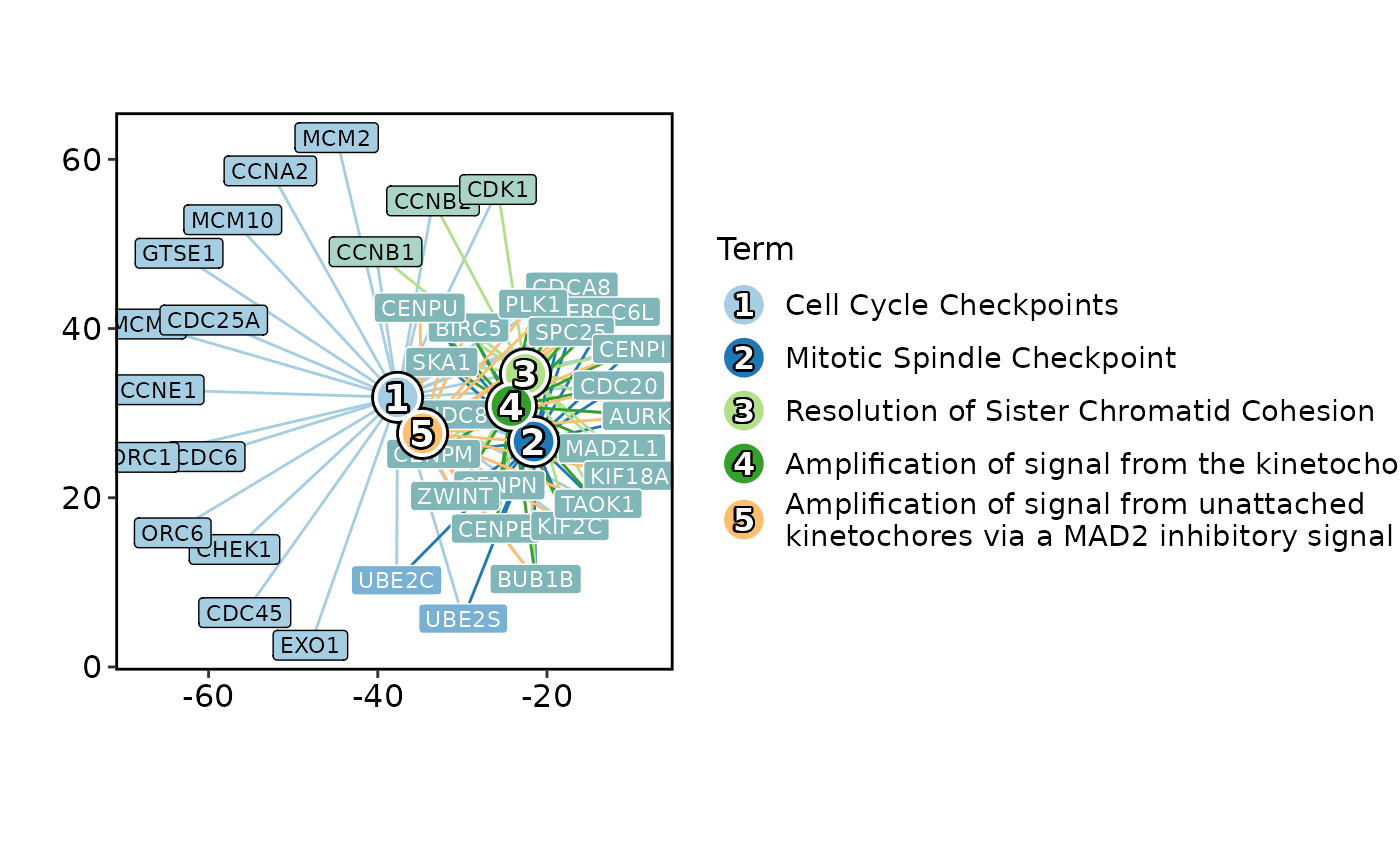 # }
# }