ScrnaMetabolicLandscape¶
Metabolic landscape analysis for scRNA-seq data
An abstract from https://github.com/LocasaleLab/Single-Cell-Metabolic-Landscape.
See also https://pwwang.github.io/biopipen/pipelines/scrna_metabolic/.
This is a group of processes to analyze the metabolic landscape of single cell RNA-seq data.
It collects a set of processes and owns a set of arguments. These arguments could either preset the default values for the processes or define the relationships between the processes.
Processes¶
The processes in this group implement part of the pipeline below from the original paper.
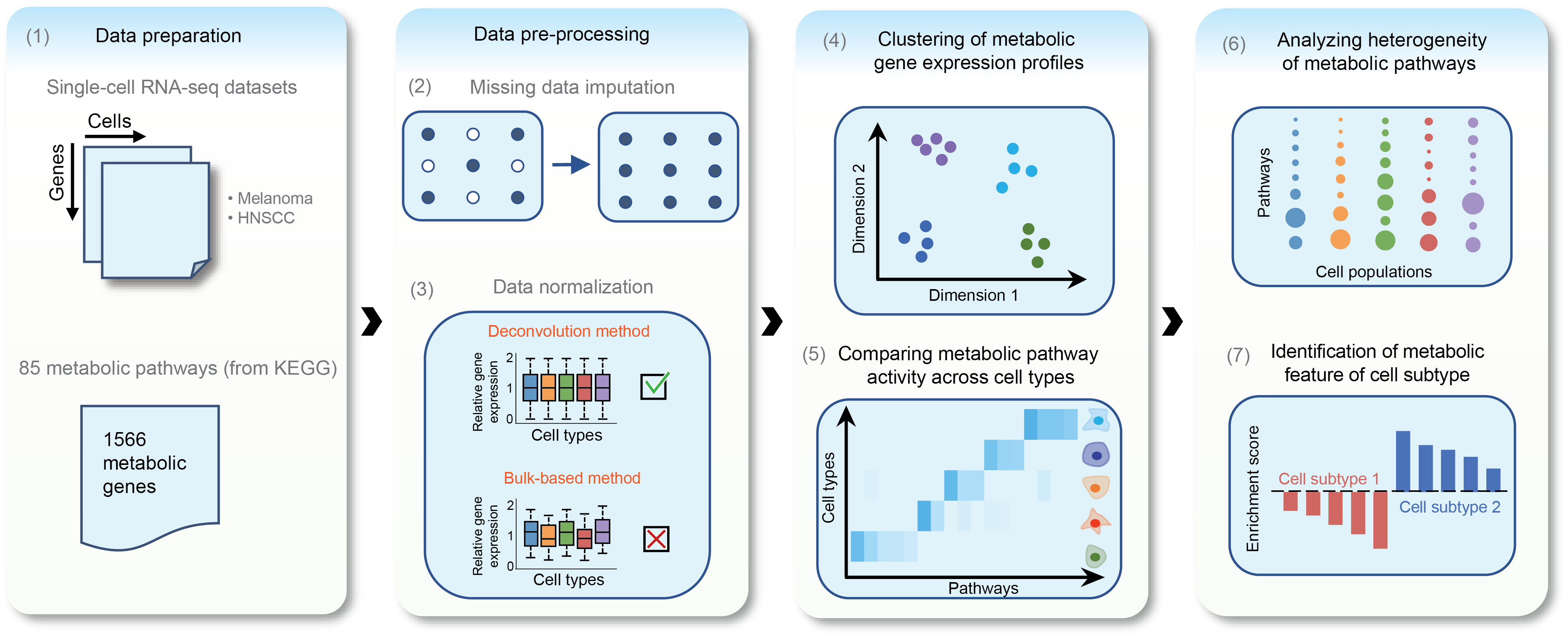
The data preparation, preprocessing and clustering already done by other processes of this pipeline. The processes in this group are used to analyze the metabolic landscape of the data.
MetabolicInput: Input for the metabolic pathway analysis pipeline for scRNA-seq dataMetabolicExprImputation: Impute the missing values in the expression dataMetabolicPathwayActivity: Calculate the pathway activities for each groupMetabolicPathwayHeterogeneity: Calculate the pathway heterogeneityMetabolicFeatures: Inter-subset metabolic features - Enrichment analysis in details
Group arguments¶
noimpute(flag): Whether to do imputation for the dropouts. IfFalse, the values will be left as is.gmtfile: The GMT file with the metabolic pathways. The gene names should match the gene names in the gene list in RNAData or the Seurat object. You can also provide a URL to the GMT file. For example, from https://download.baderlab.org/EM_Genesets/current_release/Human/symbol/.subset_by(pgarg;readonly): Subset the data by the given column in the metadata. For example,Response.NAvalues will be removed in this column. If None, the data will not be subsetted.group_by(pgarg;readonly): Group the data by the given column in the metadata. For example,cluster.mutaters(type=json): Add new columns to the metadata for grouping/subsetting. They are passed tosobj@meta.data |> mutate(...). For example,{"timepoint": "if_else(treatment == 'control', 'pre', 'post')"}will add a new columntimepointto the metadata with values ofpreandpostbased on thetreatmentcolumn.ncores(type=int): Number of cores to use for parallelization for each process