MetabolicPathwayHeterogeneity¶
Calculate Metabolic Pathway heterogeneity.
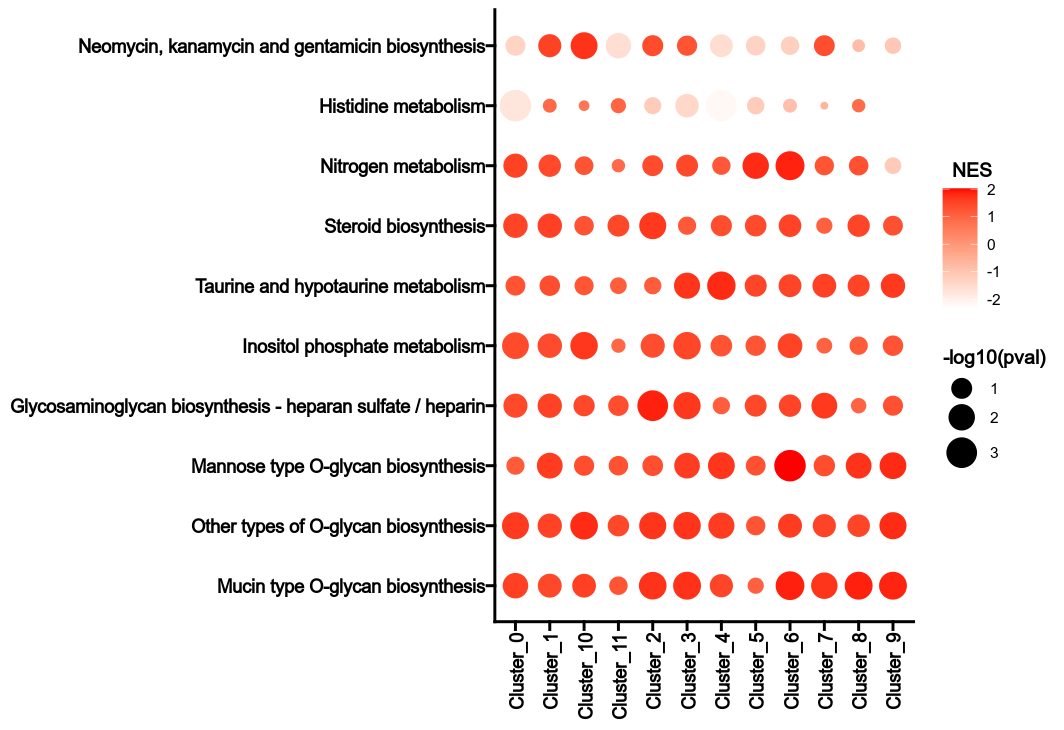
For each subset, the normalized enrichment score (NES) of each metabolic pathway
is calculated for each group.
The NES is calculated by comparing the enrichment score of the subset to the
enrichment scores of the same subset in the permutations.
The p-value is calculated by comparing the NES to the NESs of the same subset
in the permutations.
The heterogeneity can be reflected by the NES values and the p-values in
different groups for the metabolic pathways.
Input¶
sobjfile:
Output¶
outdir: Default:{{in.sobjfile | stem}}.pathwayhetero.
Environment Variables¶
gmtfile(pgarg): The GMT file with the metabolic pathways.
Defaults toScrnaMetabolicLandscape.gmtfileselect_pcs(type=float): Default:0.8.
Select the PCs to use for the analysis.pathway_pval_cutoff(type=float): Default:0.01.
The p-value cutoff to select the enriched pathwaysncores(type=int;pgarg): Default:1.
Number of cores to use for parallelization Defaults toScrnaMetabolicLandscape.ncoressubset_by(pgarg;type=auto;readonly): Subset the data by the given column in the metadata. For example,Response.
NAvalues will be removed in this column.
Defaults toScrnaMetabolicLandscape.subset_byIf None, the data will not be subsetted.
Multiple columns can be provided as a list, which is helpful when subsets have overlapping cells.group_by(pgarg;readonly): Group the data by the given column in the metadata. For example,cluster.
Defaults toScrnaMetabolicLandscape.group_byfgsea_args(type=json): Default:{'scoreType': 'std', 'nproc': 1}.
Other arguments for thefgsea::fgsea()function.
For example,{"minSize": 15, "maxSize": 500}.
See https://rdrr.io/bioc/fgsea/man/fgsea.html for more details.plots(type=json): Default:{'Pathway Heterogeneity': Diot({'plot_type': 'dot', 'devpars': Diot({'res': 100})})}.
The plots to generate.
Names will be used as the title for the plot. Values will be the arguments passed tobiopipen.utils::VizGSEA()function.
See https://pwwang.github.io/biopipen.utils.R/reference/VizGSEA.html.cases(type=json): Default:{}.
Multiple cases for the analysis.
If you only have one case, you can specify the parameters directly toenvs.subset_by,envs.group_by,envs.fgsea_args,envs.plots,envs.select_pcs, andenvs.pathway_pval_cutoff.
The name of this default case will beenvs.subset_by.
If you have multiple cases, you can specify the parameters for each case in a dictionary. The keys will be the names of the cases and the values will be dictionaries with the parameters for each case, where the values will be inherited fromenvs.subset_by,envs.group_by,envs.fgsea_args,envs.plots,envs.select_pcs, andenvs.pathway_pval_cutoff.