Immunarch¶
Exploration of Single-cell and Bulk T-cell/Antibody Immune Repertoires
Changed in 0.10.0
envs.mutaters are now applied at cell level.
Seurat clustering information and other information are added at cell level,
which can be used to subset the cells for listed analyses.
You can now use subset to subset the cells for listed analyses, at cell level.
See https://immunarch.com/articles/web_only/v3_basic_analysis.html
After ImmunarchLoading loads the raw data into an immunarch object,
this process wraps the functions from immunarch to do the following:
- Basic statistics, provided by
immunarch::repExplore, such as number of clones or distributions of lengths and counts. - The clonality of repertoires, provided by
immunarch::repClonality - The repertoire overlap, provided by
immunarch::repOverlap - The repertoire overlap, including different clustering procedures and PCA, provided by
immunarch::repOverlapAnalysis - The distributions of V or J genes, provided by
immunarch::geneUsage - The diversity of repertoires, provided by
immunarch::repDiversity - The dynamics of repertoires across time points/samples, provided by
immunarch::trackClonotypes - The spectratype of clonotypes, provided by
immunarch::spectratype - The distributions of kmers and sequence profiles, provided by
immunarch::getKmers - The V-J junction circos plots, implemented within the script of this process.
Input¶
immdata: The data loaded byimmunarch::repLoad()metafile: A cell-level metafile, where the first column must be the cell barcodes that match the cell barcodes inimmdata. The other columns can be any metadata that you want to use for the analysis. The loaded metadata will be left-joined to the converted cell-level data fromimmdata.
This can also be a Seurat object RDS file. If so, thesobj@meta.datawill be used as the metadata.
Output¶
outdir: Default:{{in.immdata | stem}}.immunarch.
The output directory
Environment Variables¶
mutaters(type=json;order=-9): Default:{}.
The mutaters passed todplyr::mutate()on expanded cell-level data to add new columns.
The keys will be the names of the columns, and the values will be the expressions.
The new names can be used involumes,lens,counts,top_clones,rare_clones,hom_clones,gene_usages,divs, etc.prefix: The prefix to the barcodes. You can use placeholder like{Sample}_The prefixed barcodes will be used to match the barcodes inin.metafile.
Not used ifin.metafileis not specified.
IfNone(default),immdata$prefixwill be used.volumes(ns): Explore clonotype volume (sizes).by: Groupings when visualize clonotype volumes, passed to the.byargument ofvis(imm_vol, .by = <values>).
Multiple columns should be separated by,.devpars(ns): The parameters for the plotting device.width(type=int): Default:1000.
The width of the plot.height(type=int): Default:1000.
The height of the plot.res(type=int): Default:100.
The resolution of the plot.
subset: Subset the data before calculating the clonotype volumes.
The whole data will be expanded to cell level, and then subsetted.
Clone sizes will be re-calculated based on the subsetted data.cases(type=json;order=9): Default:{}.
If you have multiple cases, you can use this argument to specify them.
The keys will be the names of the cases.
The values will be passed to the corresponding arguments above.
If any of these arguments are not specified, the values inenvs.volumeswill be used.
If NO cases are specified, the default case will be added, with the nameDEFAULTand the values ofenvs.volume.by,envs.volume.devpars.
lens(ns): Explore clonotype CDR3 lengths.by: Groupings when visualize clonotype lengths, passed to the.byargument ofvis(imm_len, .by = <values>).
Multiple columns should be separated by,.devpars(ns): The parameters for the plotting device.width(type=int): Default:1000.
The width of the plot.height(type=int): Default:1000.
The height of the plot.res(type=int): Default:100.
The resolution of the plot.
subset: Subset the data before calculating the clonotype volumes.
The whole data will be expanded to cell level, and then subsetted.
Clone sizes will be re-calculated based on the subsetted data.cases(type=json;order=9): Default:{}.
If you have multiple cases, you can use this argument to specify them.
The keys will be the names of the cases.
The values will be passed to the corresponding arguments above.
If any of these arguments are not specified, the values inenvs.lenswill be used.
If NO cases are specified, the default case will be added, with the nameDEFAULTand the values ofenvs.lens.by,envs.lens.devpars.
counts(ns): Explore clonotype counts.by: Groupings when visualize clonotype counts, passed to the.byargument ofvis(imm_count, .by = <values>).
Multiple columns should be separated by,.devpars(ns): The parameters for the plotting device.width(type=int): Default:1000.
The width of the plot.height(type=int): Default:1000.
The height of the plot.res(type=int): Default:100.
The resolution of the plot.
subset: Subset the data before calculating the clonotype volumes.
The whole data will be expanded to cell level, and then subsetted.
Clone sizes will be re-calculated based on the subsetted data.cases(type=json;order=9): Default:{}.
If you have multiple cases, you can use this argument to specify them.
The keys will be the names of the cases.
The values will be passed to the corresponding arguments above.
If any of these arguments are not specified, the values inenvs.countswill be used.
If NO cases are specified, the default case will be added, with the nameDEFAULTand the values ofenvs.counts.by,envs.counts.devpars.
top_clones(ns): Explore top clonotypes.by: Groupings when visualize top clones, passed to the.byargument ofvis(imm_top, .by = <values>).
Multiple columns should be separated by,.marks(list;itype=int): Default:[10, 100, 1000, 3000, 10000, 30000, 100000.0].
A numerical vector with ranges of the top clonotypes. Passed to the.headargument ofrepClonoality().devpars(ns): The parameters for the plotting device.width(type=int): Default:1000.
The width of the plot.height(type=int): Default:1000.
The height of the plot.res(type=int): Default:100.
The resolution of the plot.
subset: Subset the data before calculating the clonotype volumes.
The whole data will be expanded to cell level, and then subsetted.
Clone sizes will be re-calculated based on the subsetted data.cases(type=json;order=9): Default:{}.
If you have multiple cases, you can use this argument to specify them.
The keys will be the names of the cases.
The values will be passed to the corresponding arguments above.
If any of these arguments are not specified, the values inenvs.top_cloneswill be used.
If NO cases are specified, the default case will be added, with the nameDEFAULTand the values ofenvs.top_clones.by,envs.top_clones.marksandenvs.top_clones.devpars.
rare_clones(ns): Explore rare clonotypes.by: Groupings when visualize rare clones, passed to the.byargument ofvis(imm_rare, .by = <values>).
Multiple columns should be separated by,.marks(list;itype=int): Default:[1, 3, 10, 30, 100].
A numerical vector with ranges of abundance for the rare clonotypes in the dataset.
Passed to the.boundargument ofrepClonoality().devpars(ns): The parameters for the plotting device.width(type=int): Default:1000.
The width of the plot.height(type=int): Default:1000.
The height of the plot.res(type=int): Default:100.
The resolution of the plot.
subset: Subset the data before calculating the clonotype volumes.
The whole data will be expanded to cell level, and then subsetted.
Clone sizes will be re-calculated based on the subsetted data.cases(type=json;order=9): Default:{}.
If you have multiple cases, you can use this argument to specify them.
The keys will be the names of the cases.
The values will be passed to the corresponding arguments above.
If any of these arguments are not specified, the values inenvs.rare_cloneswill be used.
If NO cases are specified, the default case will be added, with the nameDEFAULTand the values ofenvs.rare_clones.by,envs.rare_clones.marksandenvs.rare_clones.devpars.
hom_clones(ns): Explore homeo clonotypes.by: Groupings when visualize homeo clones, passed to the.byargument ofvis(imm_hom, .by = <values>).
Multiple columns should be separated by,.marks(ns): A dict with the threshold of the half-closed intervals that mark off clonal groups.
Passed to the.clone.typesarguments ofrepClonoality().
The keys could be:Rare(type=float): Default:1e-05.
the rare clonotypesSmall(type=float): Default:0.0001.
the small clonotypesMedium(type=float): Default:0.001.
the medium clonotypesLarge(type=float): Default:0.01.
the large clonotypesHyperexpanded(type=float): Default:1.0.
the hyperexpanded clonotypes
subset: Subset the data before calculating the clonotype volumes.
The whole data will be expanded to cell level, and then subsetted.
Clone sizes will be re-calculated based on the subsetted data.devpars(ns): The parameters for the plotting device.width(type=int): Default:1000.
The width of the plot.height(type=int): Default:1000.
The height of the plot.res(type=int): Default:100.
The resolution of the plot.
cases(type=json;order=9): Default:{}.
If you have multiple cases, you can use this argument to specify them.
The keys will be the names of the cases.
The values will be passed to the corresponding arguments above.
If any of these arguments are not specified, the values inenvs.hom_cloneswill be used.
If NO cases are specified, the default case will be added, with the nameDEFAULTand the values ofenvs.hom_clones.by,envs.hom_clones.marksandenvs.hom_clones.devpars.
overlaps(ns): Explore clonotype overlaps.method(choice): Default:public.
The method to calculate overlaps.public: number of public clonotypes between two samples.overlap: a normalised measure of overlap similarity.
It is defined as the size of the intersection divided by the smaller of the size of the two sets.jaccard: conceptually a percentage of how many objects two sets have in common out of how many objects they have total.tversky: an asymmetric similarity measure on sets that compares a variant to a prototype.cosine: a measure of similarity between two non-zero vectors of an inner product space that measures the cosine of the angle between them.morisita: how many times it is more likely to randomly select two sampled points from the same quadrat (the dataset is covered by a regular grid of changing size) then it would be in the case of a random distribution generated from a Poisson process. Duplicate objects are merged with their counts are summed up.inc+public: incremental overlaps of the N most abundant clonotypes with incrementally growing N using the public method.inc+morisita: incremental overlaps of the N most abundant clonotypes with incrementally growing N using the morisita method.
subset: Subset the data before calculating the clonotype volumes.
The whole data will be expanded to cell level, and then subsetted.
Clone sizes will be re-calculated based on the subsetted data.vis_args(type=json): Default:{}.
Other arguments for the plotting functionsvis(imm_ov, ...).devpars(ns): The parameters for the plotting device.width(type=int): Default:1000.
The width of the plot.height(type=int): Default:1000.
The height of the plot.res(type=int): Default:100.
The resolution of the plot.
analyses(ns;order=8): Perform overlap analyses.method: Default:none.
Plot the samples with these dimension reduction methods.
The methods could behclust,tsne,mdsor combination of them, such asmds+hclust.
You can also set tononeto skip the analyses.
They could also be combined, for example,mds+hclust.
See https://immunarch.com/reference/repOverlapAnalysis.html.vis_args(type=json): Default:{}.
Other arguments for the plotting functions.devpars(ns): The parameters for the plotting device.width(type=int): Default:1000.
The width of the plot.height(type=int): Default:1000.
The height of the plot.res(type=int): Default:100.
The resolution of the plot.
cases(type=json): Default:{}.
If you have multiple cases, you can use this argument to specify them.
The keys will be the names of the cases.
The values will be passed to the corresponding arguments above.
If any of these arguments are not specified, the values inenvs.overlaps.analyseswill be used.
If NO cases are specified, the default case will be added, with the nameDEFAULTand the values ofenvs.overlaps.analyses.method,envs.overlaps.analyses.vis_argsandenvs.overlaps.analyses.devpars.
cases(type=json;order=9): Default:{}.
If you have multiple cases, you can use this argument to specify them.
The keys will be the names of the cases.
The values will be passed to the corresponding arguments above.
If any of these arguments are not specified, the values inenvs.overlapswill be used.
If NO cases are specified, the default case will be added, with the key the default method and the values ofenvs.overlaps.method,envs.overlaps.vis_args,envs.overlaps.devparsandenvs.overlaps.analyses.
gene_usages(ns): Explore gene usages.top(type=int): Default:30.
How many top (ranked by total usage across samples) genes to show in the plots.
Use0to use all genes.norm(flag): Default:False.
If True then use proportions of genes, else use counts of genes.by: Groupings to show gene usages, passed to the.byargument ofvis(imm_gu_top, .by = <values>).
Multiple columns should be separated by,.vis_args(type=json): Default:{}.
Other arguments for the plotting functions.devpars(ns): The parameters for the plotting device.width(type=int): Default:1000.
The width of the plot.height(type=int): Default:1000.
The height of the plot.res(type=int): Default:100.
The resolution of the plot.
subset: Subset the data before calculating the clonotype volumes.
The whole data will be expanded to cell level, and then subsetted.
Clone sizes will be re-calculated based on the subsetted data.analyses(ns;order=8): Perform gene usage analyses.method: Default:none.
The method to control how the data is going to be preprocessed and analysed.
One ofjs,cor,cosine,pca,mdsandtsne. Can also be combined with following methods for the actual analyses:hclust,kmeans,dbscan, andkruskal. For example:cosine+hclust.
You can also set tononeto skip the analyses.
See https://immunarch.com/articles/web_only/v5_gene_usage.html.vis_args(type=json): Default:{}.
Other arguments for the plotting functions.devpars(ns): The parameters for the plotting device.width(type=int): Default:1000.
The width of the plot.height(type=int): Default:1000.
The height of the plot.res(type=int): Default:100.
The resolution of the plot.
cases(type=json): Default:{}.
If you have multiple cases, you can use this argument to specify them.
The keys will be the names of the cases.
The values will be passed to the corresponding arguments above.
If any of these arguments are not specified, the values inenvs.gene_usages.analyseswill be used.
If NO cases are specified, the default case will be added, with the nameDEFAULTand the values ofenvs.gene_usages.analyses.method,envs.gene_usages.analyses.vis_argsandenvs.gene_usages.analyses.devpars.
cases(type=json;order=9): Default:{}.
If you have multiple cases, you can use this argument to specify them.
The keys will be used as the names of the cases.
The values will be passed to the corresponding arguments above.
If any of these arguments are not specified, the values inenvs.gene_usageswill be used.
If NO cases are specified, the default case will be added, with the nameDEFAULTand the values ofenvs.gene_usages.top,envs.gene_usages.norm,envs.gene_usages.by,envs.gene_usages.vis_args,envs.gene_usages.devparsandenvs.gene_usages.analyses.
spects(ns): Spectratyping analysis.quant: Select the column with clonal counts to evaluate.
Set toidto count every clonotype once.
Set tocountto take into the account number of clones per clonotype.
Multiple columns should be separated by,.col: A string that specifies the column(s) to be processed.
The output is one of the following strings, separated by the plus sign: "nt" for nucleotide sequences, "aa" for amino acid sequences, "v" for V gene segments, "j" for J gene segments.
E.g., pass "aa+v" for spectratyping on CDR3 amino acid sequences paired with V gene segments, i.e., in this case a unique clonotype is a pair of CDR3 amino acid and V gene segment.
Clonal counts of equal clonotypes will be summed up.subset: Subset the data before calculating the clonotype volumes.
The whole data will be expanded to cell level, and then subsetted.
Clone sizes will be re-calculated based on the subsetted data.devpars(ns): The parameters for the plotting device.width(type=int): Default:1000.
The width of the plot.height(type=int): Default:1000.
The height of the plot.res(type=int): Default:100.
The resolution of the plot.
cases(type=json;order=9): Default:{'By_Clonotype': Diot({'quant': 'id', 'col': 'nt'}), 'By_Num_Clones': Diot({'quant': 'count', 'col': 'aa+v'})}.
If you have multiple cases, you can use this argument to specify them.
The keys will be the names of the cases.
The values will be passed to the corresponding arguments above.
If any of these arguments are not specified, the values inenvs.spectswill be used.
By default, aBy_Clonotypecase will be added, with the values ofquant = "id"andcol = "nt", and aBy_Num_Clonescase will be added, with the values ofquant = "count"andcol = "aa+v".
divs(ns): Parameters to control the diversity analysis.method(choice): Default:gini.
The method to calculate diversity.chao1: a nonparameteric asymptotic estimator of species richness.
(number of species in a population).hill: Hill numbers are a mathematically unified family of diversity indices.
(differing only by an exponent q).div: true diversity, or the effective number of types.
It refers to the number of equally abundant types needed for the average proportional abundance of the types to equal that observed in the dataset of interest where all types may not be equally abundant.gini.simp: The Gini-Simpson index.
It is the probability of interspecific encounter, i.e., probability that two entities represent different types.inv.simp: Inverse Simpson index.
It is the effective number of types that is obtained when the weighted arithmetic mean is used to quantify average proportional abundance of types in the dataset of interest.gini: The Gini coefficient.
It measures the inequality among values of a frequency distribution (for example levels of income).
A Gini coefficient of zero expresses perfect equality, where all values are the same (for example, where everyone has the same income).
A Gini coefficient of one (or 100 percents) expresses maximal inequality among values (for example where only one person has all the income).d50: The D50 index.
It is the number of types that are needed to cover 50%% of the total abundance.raref: Species richness from the results of sampling through extrapolation.
by: The variables (column names) to group samples.
Multiple columns should be separated by,.plot_type(choice): Default:bar.
The type of the plot, works whenbyis specified.
Not working forraref.box: Boxplotbar: Barplot with error bars
subset: Subset the data before calculating the clonotype volumes.
The whole data will be expanded to cell level, and then subsetted.
Clone sizes will be re-calculated based on the subsetted data.args(type=json): Default:{}.
Other arguments forrepDiversity().
Do not include the preceding.and use-instead of.in the argument names.
For example,do-normwill be compiled to.do.norm.
See all arguments at https://immunarch.com/reference/repDiversity.html.order(list): Default:[].
The order of the values inbyon the x-axis of the plots.
If not specified, the values will be used as-is.test(ns): Perform statistical tests between each pair of groups.
Does NOT work forraref.method(choice): Default:none.
The method to perform the testnone: No testt.test: Welch's t-testwilcox.test: Wilcoxon rank sum test
padjust(choice): Default:none.
The method to adjust p-values.
Defaults tonone.bonferroni: one-step correctionholm: step-down method using Bonferroni adjustmentshochberg: step-up method (independent)hommel: closed method based on Simes tests (non-negative)BH: Benjamini & Hochberg (non-negative)BY: Benjamini & Yekutieli (negative)fdr: Benjamini & Hochberg (non-negative)none: no correction.
separate_by: A column name used to separate the samples into different plots.split_by: A column name used to split the samples into different subplots.
Likeseparate_by, but the plots will be put in the same figure.
y-axis will be shared, even ifalign_yisFalseorymin/ymaxare not specified.
ncolwill be ignored.split_order: The order of the values insplit_byon the x-axis of the plots.
It can also be used forseparate_byto control the order of the plots.
Values can be separated by,.align_x(flag): Default:False.
Align the x-axis of multiple plots. Only works forraref.align_y(flag): Default:False.
Align the y-axis of multiple plots.ymin(type=float): The minimum value of the y-axis.
The minimum value of the y-axis for plots splitting byseparate_by.
align_yis forcedTruewhen bothyminandymaxare specified.ymax(type=float): The maximum value of the y-axis.
The maximum value of the y-axis for plots splitting byseparate_by.
align_yis forcedTruewhen bothyminandymaxare specified.
Works when bothyminandymaxare specified.log(flag): Default:False.
Indicate whether we should plot with log-transformed x-axis usingvis(.log = TRUE). Only works forraref.ncol(type=int): Default:2.
The number of columns of the plots.devpars(ns): The parameters for the plotting device.width(type=int): Default:800.
The width of the deviceheight(type=int): Default:800.
The height of the deviceres(type=int): Default:100.
The resolution of the device
cases(type=json;order=9): Default:{}.
If you have multiple cases, you can use this argument to specify them.
The keys will be used as the names of the cases.
The values will be passed to the corresponding arguments above.
If NO cases are specified, the default case will be added, with the name ofenvs.div.method.
The values specified inenvs.divwill be used as the defaults for the cases here.
trackings(ns): Parameters to control the clonotype tracking analysis.targets: Either a set of CDR3AA seq of clonotypes to track (separated by,), or simply an integer to track the top N clonotypes.subject_col: Default:Sample.
The column name in meta data that contains the subjects/samples on the x-axis of the alluvial plot.
If the values in this column are not unique, the values will be merged with the values insubject_colto form the x-axis.
This defaults toSample.subset: Subset the data before calculating the clonotype volumes.
The whole data will be expanded to cell level, and then subsetted.
Clone sizes will be re-calculated based on the subsetted data.subjects(list): Default:[].
A list of values fromsubject_colto show in the alluvial plot on the x-axis.
If not specified, all values insubject_colwill be used.
This also specifies the order of the x-axis.cases(type=json;order=9): Default:{}.
If you have multiple cases, you can use this argument to specify them.
The keys will be used as the names of the cases.
The values will be passed to the corresponding arguments (target,subject_col, andsubjects).
If any of these arguments are not specified, the values inenvs.trackingswill be used.
If NO cases are specified, the default case will be added, with the nameDEFAULTand the values ofenvs.trackings.target,envs.trackings.subject_col, andenvs.trackings.subjects.
kmers(ns): Arguments for kmer analysis.k(type=int): Default:5.
The length of kmer.head(type=int): Default:10.
The number of top kmers to show.vis_args(type=json): Default:{}.
Other arguments for the plotting functions.devpars(ns): The parameters for the plotting device.width(type=int): Default:1000.
The width of the plot.height(type=int): Default:1000.
The height of the plot.res(type=int): Default:100.
The resolution of the plot.
subset: Subset the data before calculating the clonotype volumes.
The whole data will be expanded to cell level, and then subsetted.
Clone sizes will be re-calculated based on the subsetted data.profiles(ns;order=8): Arguments for sequence profilings.method(choice): Default:self.
The method for the position matrix.
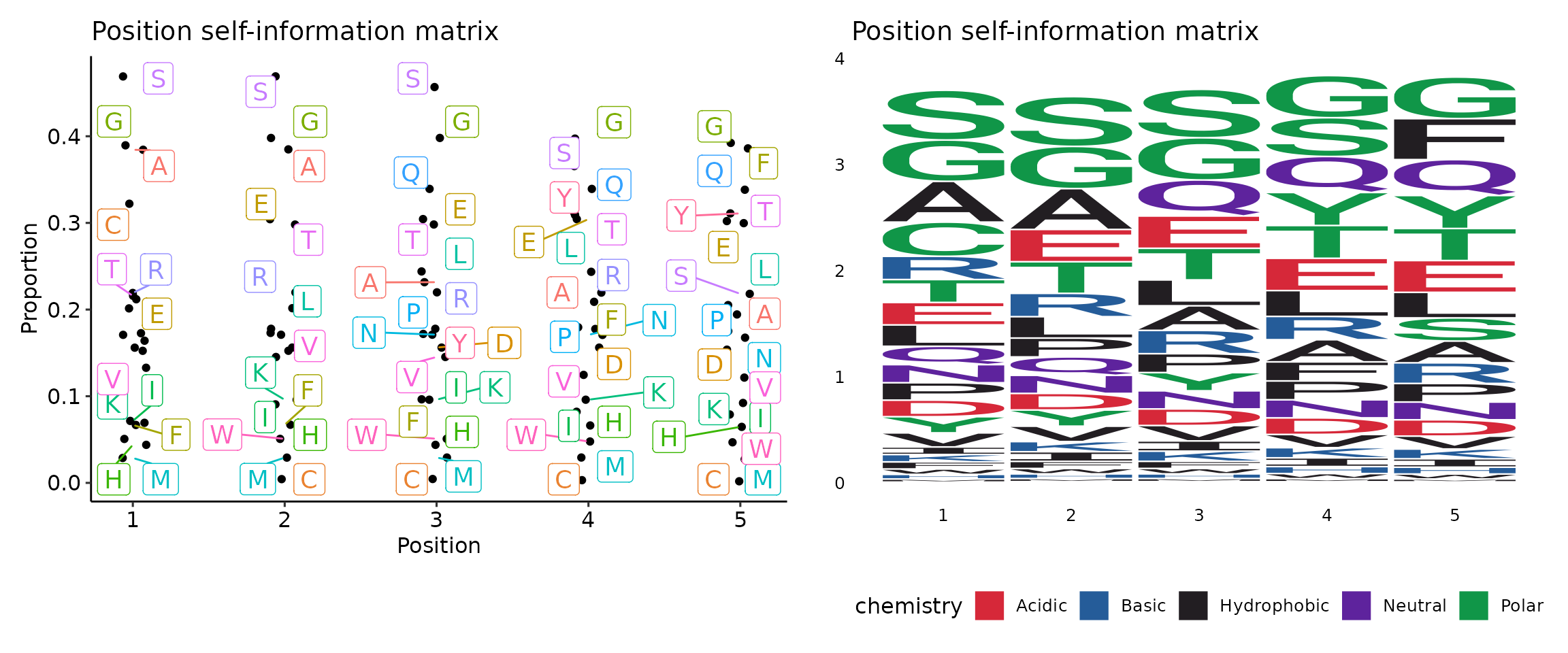
For more information see https://en.wikipedia.org/wiki/Position_weight_matrix.freq: position frequency matrix (PFM) - a matrix with occurences of each amino acid in each position.prob: position probability matrix (PPM) - a matrix with probabilities of each amino acid in each position.wei: position weight matrix (PWM) - a matrix with log likelihoods of PPM elements.self: self-information matrix (SIM) - a matrix with self-information of elements in PWM.
vis_args(type=json): Default:{}.
Other arguments for the plotting functions.devpars(ns): The parameters for the plotting device.width(type=int): Default:1000.
The width of the plot.height(type=int): Default:1000.
The height of the plot.res(type=int): Default:100.
The resolution of the plot.
cases(type=json): Default:{}.
If you have multiple cases, you can use this argument to specify them.
The keys will be the names of the cases.
The values will be passed to the corresponding arguments above.
If any of these arguments are not specified, the values inenvs.kmers.profileswill be used.
If NO cases are specified, the default case will be added, with the nameDEFAULTand the values ofenvs.kmers.profiles.method,envs.kmers.profiles.vis_argsandenvs.kmers.profiles.devpars.
cases(type=json;order=9): Default:{}.
If you have multiple cases, you can use this argument to specify them.
The keys will be used as the names of the cases.
The values will be passed to the corresponding arguments above.
If any of these arguments are not specified, the default case will be added, with the nameDEFAULTand the values ofenvs.kmers.k,envs.kmers.head,envs.kmers.vis_argsandenvs.kmers.devpars.
vj_junc(ns): Arguments for VJ junction circos plots.
This analysis is not included inimmunarch. It is a separate implementation usingcirclize.by: Default:Sample.
Groupings to show VJ usages. Typically, this is theSamplecolumn, so that the VJ usages are shown for each sample.
But you can also use other columns, such asSubjectto show the VJ usages for each subject.
Multiple columns should be separated by,.by_clones(flag): Default:True.
If True, the VJ usages will be calculated based on the distinct clonotypes, instead of the individual cells.subset: Subset the data before plotting VJ usages.
The whole data will be expanded to cell level, and then subsetted.
Clone sizes will be re-calculated based on the subsetted data, which will affect the VJ usages at cell level (by_clones=False).devpars(ns): The parameters for the plotting device.width(type=int): Default:800.
The width of the plot.height(type=int): Default:800.
The height of the plot.res(type=int): Default:100.
The resolution of the plot.
cases(type=json;order=9): Default:{}.
If you have multiple cases, you can use this argument to specify them.
The keys will be used as the names of the cases. The values will be passed to the corresponding arguments above.
If any of these arguments are not specified, the values inenvs.vj_juncwill be used.
If NO cases are specified, the default case will be added, with the nameDEFAULTand the values ofenvs.vj_junc.by,envs.vj_junc.by_clonesenvs.vj_junc.subsetandenvs.vj_junc.devpars.
Environment Variable Design¶
With different sets of arguments, a single function of the above can perform different tasks.
For example, repExplore can be used to get the statistics of the size of the repertoire,
the statistics of the length of the CDR3 region, or the statistics of the number of
the clonotypes. Other than that, you can also have different ways to visualize the results,
by passing different arguments to the immunarch::vis function.
For example, you can pass .by to vis to visualize the results of repExplore by different groups.
Before we explain each environment variable in details in the next section, we will give some examples here to show how the environment variables are organized in order for a single function to perform different tasks.
# Repertoire overlapping
[Immunarch.envs.overlaps]
# The method to calculate the overlap, passed to `repOverlap`
method = "public"
What if we want to calculate the overlap by different methods at the same time? We can use the following configuration:
[Immunarch.envs.overlaps.cases]
Public = { method = "public" }
Jaccard = { method = "jaccard" }
Then, the repOverlap function will be called twice, once with method = "public" and once with method = "jaccard". We can also use different arguments to visualize the results. These arguments will be passed to the vis function:
[Immunarch.envs.overlaps.cases.Public]
method = "public"
vis_args = { "-plot": "heatmap2" }
[Immunarch.envs.overlaps.cases.Jaccard]
method = "jaccard"
vis_args = { "-plot": "heatmap2" }
-plot will be translated to .plot and then passed to vis.
If multiple cases share the same arguments, we can use the following configuration:
[Immunarch.envs.overlaps]
vis_args = { "-plot": "heatmap2" }
[Immunarch.envs.overlaps.cases]
Public = { method = "public" }
Jaccard = { method = "jaccard" }
For some results, there are futher analysis that can be performed. For example, for the repertoire overlap, we can perform clustering and PCA (see also https://immunarch.com/articles/web_only/v4_overlap.html):
imm_ov1 <- repOverlap(immdata$data, .method = "public", .verbose = F)
repOverlapAnalysis(imm_ov1, "mds") %>% vis()
repOverlapAnalysis(imm_ov1, "tsne") %>% vis()
In such a case, we can use the following configuration:
[Immunarch.envs.overlaps]
method = "public"
[Immunarch.envs.overlaps.analyses.cases]
MDS = { "-method": "mds" }
TSNE = { "-method": "tsne" }
Then, the repOverlapAnalysis function will be called twice on the result generated by repOverlap(immdata$data, .method = "public"), once with .method = "mds" and once with .method = "tsne". We can also use different arguments to visualize the results. These arguments will be passed to the vis function:
[Immunarch.envs.overlaps]
method = "public"
[Immunarch.envs.overlaps.analyses]
# See: <https://immunarch.com/reference/vis.immunr_hclust.html>
vis_args = { "-plot": "best" }
[Immunarch.envs.overlaps.analyses.cases]
MDS = { "-method": "mds" }
TSNE = { "-method": "tsne" }
Generally, you don't need to specify cases if you only have one case. A default case will be created for you. For multiple cases, the arguments at the same level as cases will be inherited by all cases.
Examples¶
[Immunarch.envs.kmers]
k = 5
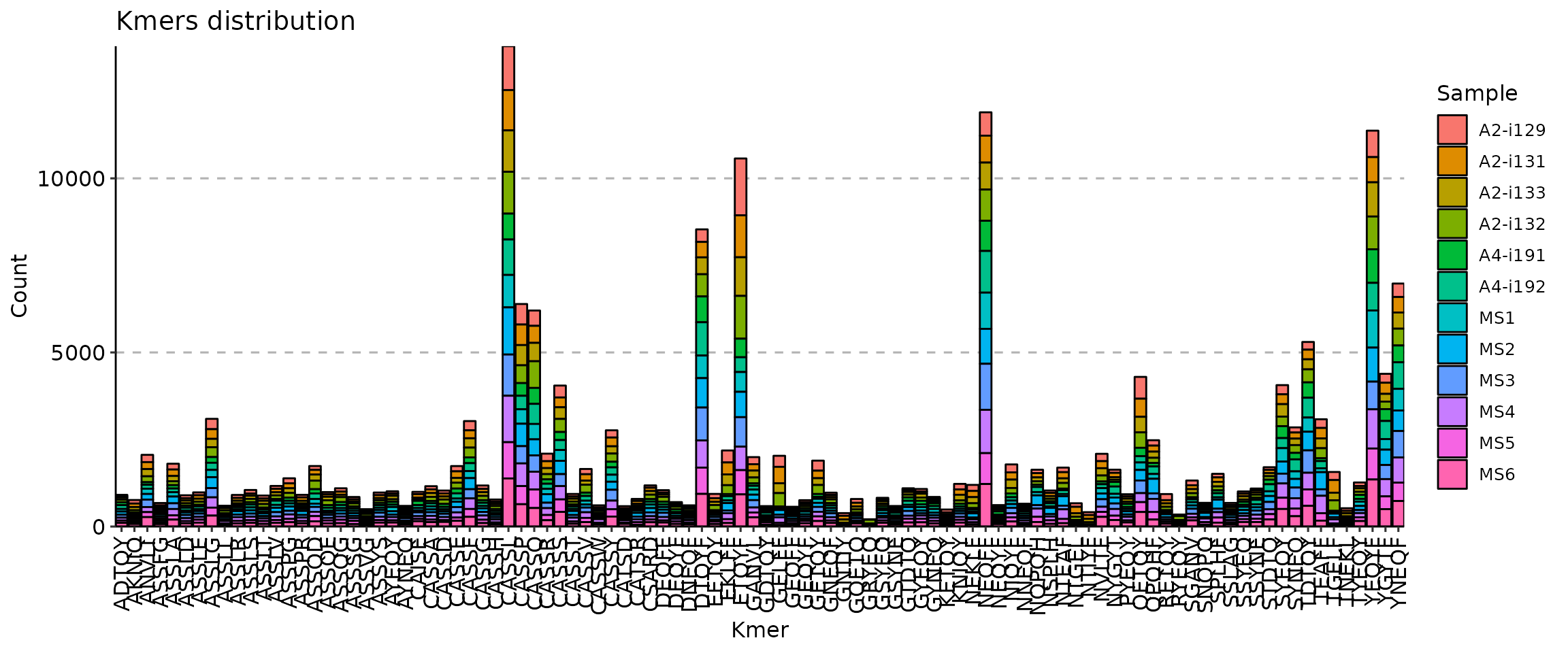
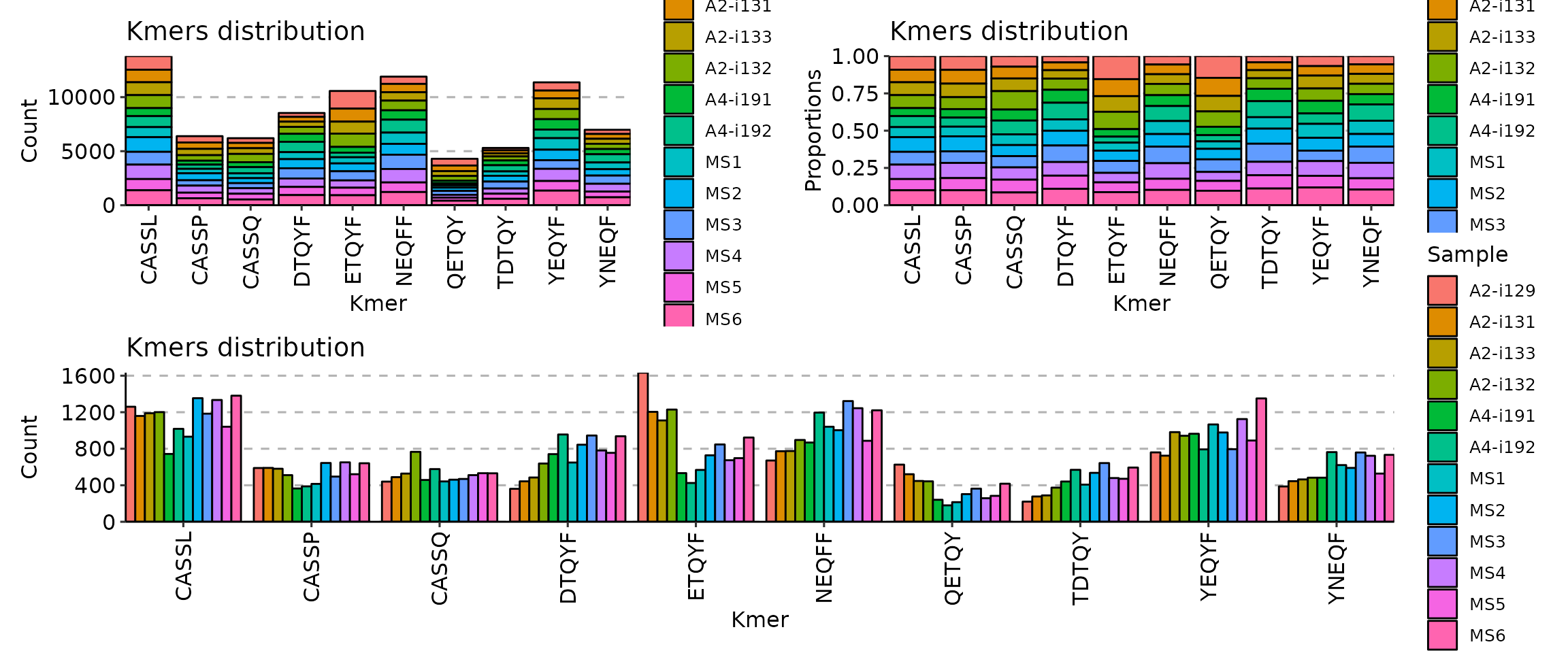
[Immunarch.envs.kmers]
# Shared by cases
k = 5
[Immunarch.envs.kmers.cases]
Head5 = { head = 5, -position = "stack" }
Head10 = { head = 10, -position = "fill" }
Head30 = { head = 30, -position = "dodge" }
With motif profiling:
[Immunarch.envs.kmers]
k = 5
[Immnuarch.envs.kmers.profiles.cases]
TextPlot = { method = "self", vis_args = { "-plot": "text" } }
SeqPlot = { method = "self", vis_args = { "-plot": "seq" } }