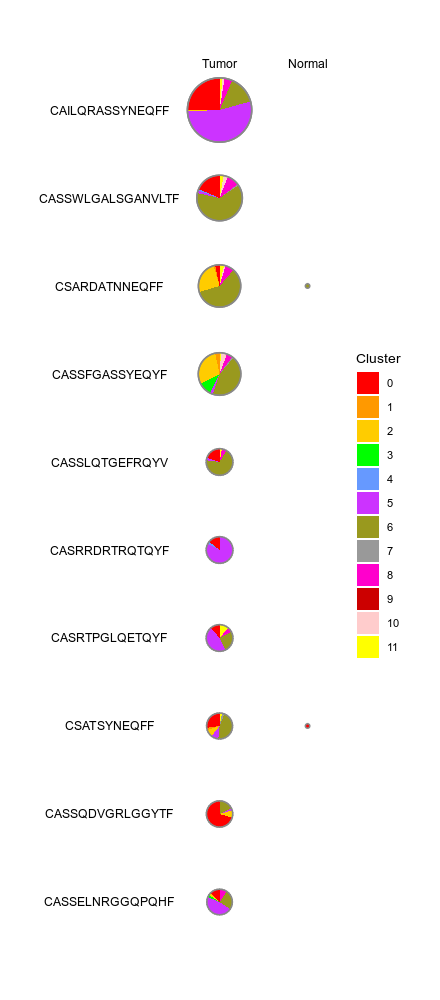
CellsDistribution¶
Distribution of cells (i.e. in a TCR clone) from different groups for each cluster
This generates a set of pie charts with proportion of cells in each cluster
Rows are the cells identities (i.e. TCR clones or TCR clusters), columns
are groups (i.e. clinic groups).
Input¶
srtobj: The seurat object in RDS format
Output¶
outdir: Default:{{in.srtobj | stem}}.cells_distribution.
The output directory
Environment Variables¶
-
mutaters(type=json): Default:{}.
The mutaters to mutate the metadata Keys are the names of the mutaters and values are the R expressions passed bydplyr::mutate()to mutate the metadata.
There are also also 4 helper functions,expanded,collapsed,emergedandvanished, which can be used to identify the expanded/collpased/emerged/vanished groups (i.e. TCR clones).
See also https://pwwang.github.io/immunopipe/configurations/#mutater-helpers.
For example, you can use{"Patient1_Tumor_Collapsed_Clones": "expanded(., Source, 'Tumor', subset = Patent == 'Patient1', uniq = FALSE)"}to create a new column in metadata namedPatient1_Tumor_Collapsed_Cloneswith the collapsed clones in the tumor sample (compared to the normal sample) of patient 1.
The values in this columns for other clones will beNA.
Those functions take following arguments:df: The metadata data frame. You can use the.to refer to it.group.by: The column name in metadata to group the cells.idents: The first group or both groups of cells to compare (value ingroup.bycolumn). If only the first group is given, the rest of the cells (with non-NA ingroup.bycolumn) will be used as the second group.subset: An expression to subset the cells, will be passed todplyr::filter(). Default isTRUE(no filtering).each: A column name (without quotes) in metadata to split the cells.
Each comparison will be done for each value in this column (typically each patient or subject).id: The column name in metadata for the group ids (i.e.CDR3.aa).compare: Either a (numeric) column name (i.e.Clones) in metadata to compare between groups, or.nto compare the number of cells in each group.
If numeric column is given, the values should be the same for all cells in the same group.
This will not be checked (only the first value is used).
It is helpful to useClonesto use the raw clone size from TCR data, in case the cells are not completely mapped to RNA data.
Also if you havesubsetset orNAs ingroup.bycolumn, you should use.nto compare the number of cells in each group.uniq: Whether to return unique ids or not. Default isTRUE. IfFALSE, you can mutate the meta data frame with the returned ids. For example,df |> mutate(expanded = expanded(...)).debug: Return the data frame with intermediate columns instead of the ids. Default isFALSE.order: The expression passed todplyr::arrange()to order intermediate dataframe and get the ids in order accordingly.
The intermediate dataframe includes the following columns:<id>: The ids of clones (i.e.CDR3.aa).<each>: The values ineachcolumn.ident_1: The size of clones in the first group.ident_2: The size of clones in the second group..diff: The difference between the sizes of clones in the first and second groups..sum: The sum of the sizes of clones in the first and second groups..predicate: Showing whether the clone is expanded/collapsed/emerged/vanished.include_emerged: Whether to include the emerged group forexpanded(only works forexpanded). Default isFALSE.include_vanished: Whether to include the vanished group forcollapsed(only works forcollapsed). Default isFALSE.
You can also use
top()to get the top clones (i.e. the clones with the largest size) in each group.
For example, you can use{"Patient1_Top10_Clones": "top(subset = Patent == 'Patient1', uniq = FALSE)"}to create a new column in metadata namedPatient1_Top10_Clones.
The values in this columns for other clones will beNA.
This function takes following arguments:
*df: The metadata data frame. You can use the.to refer to it.
*id: The column name in metadata for the group ids (i.e.CDR3.aa).
*n: The number of top clones to return. Default is10.
If n < 1, it will be treated as the percentage of the size of the group.
Specify0to get all clones.
*compare: Either a (numeric) column name (i.e.Clones) in metadata to compare between groups, or.nto compare the number of cells in each group.
If numeric column is given, the values should be the same for all cells in the same group.
This will not be checked (only the first value is used).
It is helpful to useClonesto use the raw clone size from TCR data, in case the cells are not completely mapped to RNA data.
Also if you havesubsetset orNAs ingroup.bycolumn, you should use.nto compare the number of cells in each group.
*subset: An expression to subset the cells, will be passed todplyr::filter(). Default isTRUE(no filtering).
*each: A column name (without quotes) in metadata to split the cells.
Each comparison will be done for each value in this column (typically each patient or subject).
*uniq: Whether to return unique ids or not. Default isTRUE. IfFALSE, you can mutate the meta data frame with the returned ids. For example,df |> mutate(expanded = expanded(...)).
*debug: Return the data frame with intermediate columns instead of the ids. Default isFALSE.
*with_ties: Whether to include ties (i.e. clones with the same size as the last clone) or not. Default isFALSE. -
cluster_orderby: The order of the clusters to show on the plot.
An expression passed todplyr::summarise()on the grouped data frame (byseurat_clusters).
The summary stat will be passed todplyr::arrange()to order the clusters. It's applied on the whole meta.data before grouping and subsetting.
For example, you can order the clusters by the activation score of the cluster:desc(mean(ActivationScore, na.rm = TRUE)), suppose you have a columnActivationScorein the metadata. group_by: The column name in metadata to group the cells for the columns of the plot.group_order(list): Default:[].
The order of the groups (columns) to show on the plotcells_by: The column name in metadata to group the cells for the rows of the plot.
If your cell groups have overlapping cells, you can also use multiple columns, separated by comma (,).
These columns will be concatenated to form the cell groups. For the overlapping cells, they will be counted multiple times for different groups. So make sure the cell group names in different columns are unique.cells_order(list): Default:[].
The order of the cells (rows) to show on the plotcells_orderby: An expression passed todplyr::arrange()to order the cells (rows) of the plot.
Only works whencells-orderis not specified.
The data frame passed todplyr::arrange()is grouped bycells_bybefore ordering.
You can have multiple expressions separated by semicolon (;). The expessions will be parsed byrlang::parse_exprs().
4 extra columns were added to the metadata for ordering the rows in the plot:CloneSize: The size (number of cells) of clones (identified bycells_by)CloneGroupSize: The clone size in each group (identified bygroup_by)CloneClusterSize: The clone size in each cluster (identified byseurat_clusters)CloneGroupClusterSize: The clone size in each group and cluster (identified bygroup_byandseurat_clusters)
cells_n(type=int): Default:10.
The max number of groups to show for each cell group identity (row).
Ignored ifcells_orderis specified.subset: An expression to subset the cells, will be passed todplyr::filter()on metadata.
This will be applied prior toeach.descr: The description of the case, will be shown in the report.hm_devpars(ns): The device parameters for the heatmaps.res(type=int): The resolution of the heatmaps.height(type=int): The height of the heatmaps.width(type=int): The width of the heatmaps.
devpars(ns): The device parameters for the plots of pie charts.res(type=int): The resolution of the plotsheight(type=int): The height of the plotswidth(type=int): The width of the plots
each: The column name in metadata to separate the cells into different plots.prefix_each(flag): Default:True.
Whether to prefix theeachcolumn name to the value as the case/section name.section: Default:DEFAULT.
The section to show in the report. This allows different cases to be put in the same section in report.
Only works wheneachis not specified.
Thesectionis used to collect cases and put the results under the same directory and the same section in report.
Wheneachfor a case is specified, thesectionwill be ignored and case name will be used assection.
The cases will be the expanded values ineachcolumn. Whenprefix_eachis True, the column name specified byeachwill be prefixed to each value as directory name and expanded case name.overlap(list): Default:[].
Plot the overlap of cell groups (values ofcells_by) in different cases under the same section.
The section must have at least 2 cases, each case should have a singlecells_bycolumn.cases(type=json;order=99): Default:{}.
If you have multiple cases, you can specify them here.
Keys are the names of the cases and values are the options above exceptmutaters.
If some options are not specified, the options inenvswill be used.
If no cases are specified, a default case will be used with case nameDEFAULT.
Examples¶
[CellsDistribution.envs.mutaters]
# Add Patient1_Tumor_Expanded column with CDR3.aa that
# expands in Tumor of patient 1
Patient1_Tumor_Expanded = '''
expanded(., region, "Tumor", subset = patient == "Lung1", uniq = FALSE)
'''
[CellsDistribution.envs.cases.Patient1_Tumor_Expanded]
cells_by = "Patient1_Tumor_Expanded"
cells_orderby = "desc(CloneSize)"
group_by = "region"
group_order = [ "Tumor", "Normal" ]