"""Metabolic landscape analysis for scRNA-seq data"""
from __future__ import annotations
from pathlib import Path
from typing import Type
from diot import Diot # type: ignore
from datar.tibble import tibble
from pipen.utils import mark
from pipen_args import ProcGroup
from pipen_annotate import annotate
from ..core.config import config
from ..core.proc import Proc
class MetabolicPathwayActivity(Proc):DOCS
"""This process calculates the pathway activities in different groups and subsets.
The cells are first grouped by subsets and then the metabolic activities are
examined for each groups in different subsets.
For each subset, a heatmap and a violin plot will be generated.
The heatmap shows the pathway activities for each group and each metabolic pathway
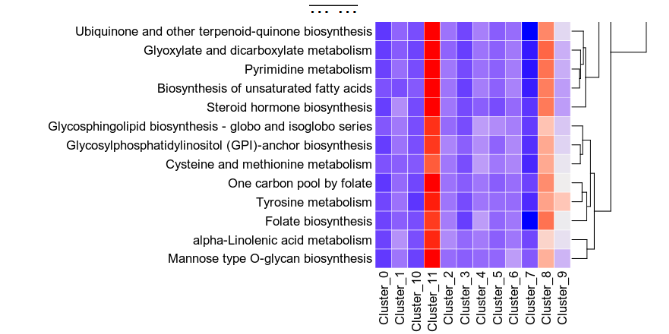{: width="80%"}
The violin plot shows the distribution of the pathway activities for each group
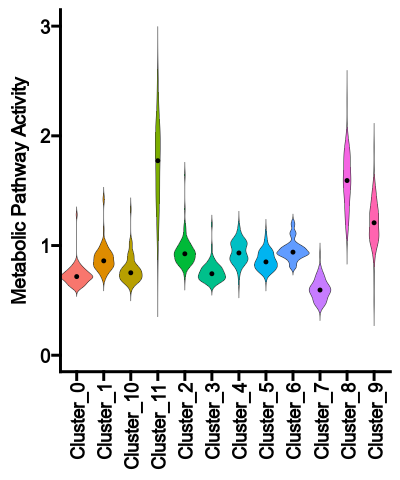{: width="45%"}
You may also have a merged heatmap to show all subsets in one plot.
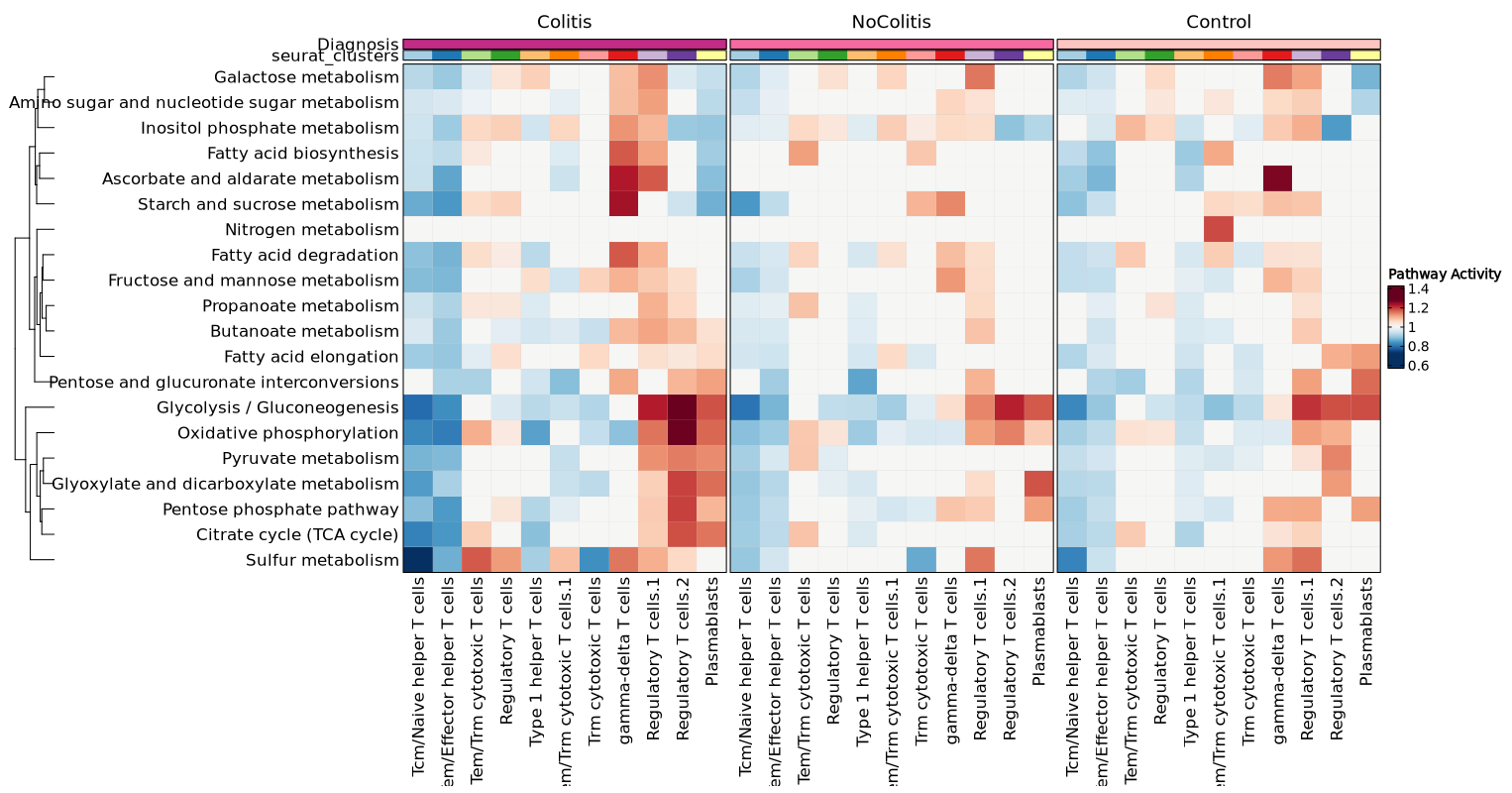{: width="80%"}
Input:
sobjfile: The Seurat object file.
It should be loaded as a Seurat object
Output:
outdir: The output directory.
It will contain the pathway activity score files and plots.
Envs:
ntimes (type=int): Number of permutations to estimate the p-values
ncores (type=int;pgarg): Number of cores to use for parallelization
Defaults to `ScrnaMetabolicLandscape.ncores`
gmtfile (pgarg): The GMT file with the metabolic pathways.
Defaults to `ScrnaMetabolicLandscape.gmtfile`
subset_by (pgarg;type=auto;readonly): Subset the data by the given column in the
metadata. For example, `Response`.
`NA` values will be removed in this column.
Defaults to `ScrnaMetabolicLandscape.subset_by`
If None, the data will not be subsetted.
Multiple columns can be provided as a list, which is helpful when
subsets have overlapping cells.
group_by (pgarg;readonly): Group the data by the given column in the
metadata. For example, `cluster`.
Defaults to `ScrnaMetabolicLandscape.group_by`
plots (type=json): The plots to generate.
Names will be used as the prefix for the output files. Values will be
a dictionary with the following keys:
* `plot_type` is the type of plot to generate. One of `heatmap`,
`box`, `violin` or `merged_heatmap` (all subsets in one plot).
* `devpars` is a dictionary with the device parameters for the plot.
* Other arguments for `plotthis::Heatmap()`, `plotthis::BoxPlot()`
or `plotthis::ViolinPlot()`, depending on the `plot_type`.
cases (type=json): Multiple cases for the analysis.
If you only have one case, you can specify the parameters directly to
`envs.ntimes`, `envs.subset_by`, `envs.group_by`, `envs.group1`,
`envs.group2`, and `envs.plots`. The name of the case will be
`envs.subset_by`.
If you have multiple cases, you can specify the parameters for each case
in a dictionary. The keys will be the names of the cases and the values
will be dictionaries with the parameters for each case, where the values
will be inherited from `envs.ntimes`, `envs.subset_by`, `envs.group_by`,
`envs.group1`, `envs.group2`, and `envs.plots`.
""" # noqa: E501
input = "sobjfile:file"
output = "outdir:dir:{{in.sobjfile | stem}}.pathwayactivity"
envs = {
"ntimes": 5000,
"ncores": config.misc.ncores,
"gmtfile": None,
"subset_by": None,
"group_by": None,
"plots": {
"Pathway Activity (violin plot)": {
"plot_type": "violin",
"add_box": True,
"devpars": {"res": 100},
},
"Pathway Activity (heatmap)": {
"plot_type": "heatmap",
"devpars": {"res": 100},
},
},
"cases": {},
}
lang = config.lang.rscript
script = (
"file://../scripts/scrna_metabolic_landscape/MetabolicPathwayActivity.R"
)
plugin_opts = {
"report":
"file://../reports/scrna_metabolic_landscape/MetabolicPathwayActivity.svelte"
}
class MetabolicFeatures(Proc):DOCS
"""This process performs enrichment analysis for the metabolic pathways
for each group in each subset.
The enrichment analysis is done with [`fgsea`](https://bioconductor.org/packages/release/bioc/html/fgsea.html)
package or the [`GSEA_R`](https://github.com/GSEA-MSigDB/GSEA_R) package.
Input:
sobjfile: The Seurat object file in rds.
It should be loaded as a Seurat object
Output:
outdir: The output directory.
It will contain the GSEA results and plots.
Envs:
ncores (type=int;pgarg): Number of cores to use for parallelization for
the comparisons for each subset and group.
Defaults to `ScrnaMetabolicLandscape.ncores`.
prerank_method (choice): Method to use for gene preranking.
Signal to noise: the larger the differences of the means
(scaled by the standard deviations); that is, the more distinct
the gene expression is in each phenotype and the more the gene
acts as a “class marker.”.
Absolute signal to noise: the absolute value of the signal to
noise.
T test: Uses the difference of means scaled by the standard
deviation and number of samples.
Ratio of classes: Uses the ratio of class means to calculate
fold change for natural scale data.
Diff of classes: Uses the difference of class means to calculate
fold change for nature scale data
Log2 ratio of classes: Uses the log2 ratio of class means to
calculate fold change for natural scale data. This is the
recommended statistic for calculating fold change for log scale
data.
- signal_to_noise: Signal to noise
- s2n: Alias of signal_to_noise
- abs_signal_to_noise: absolute signal to noise
- abs_s2n: Alias of abs_signal_to_noise
- t_test: T test
- ratio_of_classes: Also referred to as fold change
- diff_of_classes: Difference of class means
- log2_ratio_of_classes: Log2 ratio of class means
gmtfile (pgarg): The GMT file with the metabolic pathways.
Defaults to `ScrnaMetabolicLandscape.gmtfile`
subset_by (pgarg;type=auto;readonly): Subset the data by the given column in the
metadata. For example, `Response`.
`NA` values will be removed in this column.
Defaults to `ScrnaMetabolicLandscape.subset_by`
If None, the data will not be subsetted.
Multiple columns can be provided as a list, which is helpful when
subsets have overlapping cells.
group_by (pgarg;readonly): Group the data by the given column in the
metadata. For example, `cluster`.
Defaults to `ScrnaMetabolicLandscape.group_by`
comparisons (type=list): The comparison groups to use for the analysis.
If not provided, each group in the `group_by` column will be used
to compare with the other groups.
If a single group is provided as an element, it will be used to
compare with all the other groups.
For example, if we have `group_by = "cluster"` and we have
`1`, `2` and `3` in the `group_by` column, we could have
`comparisons = ["1", "2"]`, which will compare the group `1` with groups
`2` and `3`, and the group `2` with groups `1` and `3`. We could also
have `comparisons = ["1:2", "1:3"]`, which will compare the group `1` with
group `2` and group `1` with group `3`.
fgsea_args (type=json): Other arguments for the `fgsea::fgsea()` function.
For example, `{"minSize": 15, "maxSize": 500}`.
See <https://rdrr.io/bioc/fgsea/man/fgsea.html> for more details.
plots (type=json): The plots to generate.
Names will be used as the title for the plot. Values will be the arguments
passed to `biopipen.utils::VizGSEA()` function.
See <https://pwwang.github.io/biopipen.utils.R/reference/VizGSEA.html>.
A key `level` is supported to specify the level of the plot.
Possible values are `case`, which includes all subsets and groups in the
case; `subset`, which includes all groups in the subset; otherwise, it
will plot for the groups.
For `case`/`subset` level plots, current `plot_type` only "dot" is supported
for now, then the values will be passed to `plotthis::DotPlot()`
cases (type=json): Multiple cases for the analysis.
If you only have one case, you can specify the parameters directly to
`envs.prerank_method`, `envs.subset_by`, `envs.group_by`,
`envs.comparisons`, `envs.fgsea_args` and `envs.plots`.
The name of this default case will be `envs.subset_by`.
If you have multiple cases, you can specify the parameters for each case
in a dictionary. The keys will be the names of the cases and the values
will be dictionaries with the parameters for each case, where the values
will be inherited from `envs.prerank_method`,
`envs.subset_by`, `envs.group_by`, `envs.comparisons`, `envs.fgsea_args`
and `envs.plots`.
""" # noqa: E501
input = "sobjfile:file"
output = "outdir:dir:{{in.sobjfile | stem}}.pathwayfeatures"
lang = config.lang.rscript
envs = {
"ncores": config.misc.ncores,
"prerank_method": "signal_to_noise",
"gmtfile": None,
"subset_by": None,
"group_by": None,
"comparisons": [],
"fgsea_args": {},
"plots": {
"Summary Plot": {
"plot_type": "summary",
"top_term": 10,
"devpars": {"res": 100},
},
"Enrichment Plots": {
"plot_type": "gsea",
"top_term": 10,
"devpars": {"res": 100},
},
},
"cases": {},
}
script = "file://../scripts/scrna_metabolic_landscape/MetabolicFeatures.R"
plugin_opts = {
"report": "file://../reports/scrna_metabolic_landscape/MetabolicFeatures.svelte"
}
class MetabolicPathwayHeterogeneity(Proc):DOCS
"""Calculate Metabolic Pathway heterogeneity.
For each subset, the normalized enrichment score (NES) of each metabolic pathway
is calculated for each group.
The NES is calculated by comparing the enrichment score of the subset to the
enrichment scores of the same subset in the permutations.
The p-value is calculated by comparing the NES to the NESs of the same subset
in the permutations.
The heterogeneity can be reflected by the NES values and the p-values in
different groups for the metabolic pathways.
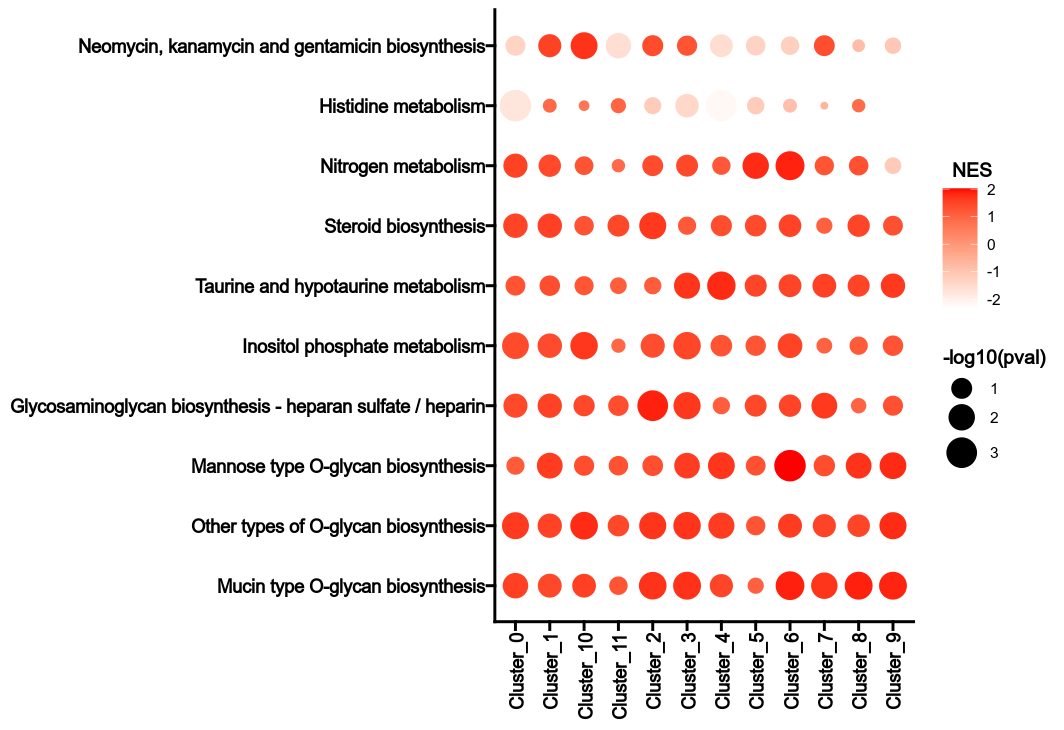
Envs:
gmtfile (pgarg): The GMT file with the metabolic pathways.
Defaults to `ScrnaMetabolicLandscape.gmtfile`
select_pcs (type=float): Select the PCs to use for the analysis.
pathway_pval_cutoff (type=float): The p-value cutoff to select
the enriched pathways
ncores (type=int;pgarg): Number of cores to use for parallelization
Defaults to `ScrnaMetabolicLandscape.ncores`
subset_by (pgarg;type=auto;readonly): Subset the data by the given column in the
metadata. For example, `Response`.
`NA` values will be removed in this column.
Defaults to `ScrnaMetabolicLandscape.subset_by`
If None, the data will not be subsetted.
Multiple columns can be provided as a list, which is helpful when
subsets have overlapping cells.
group_by (pgarg;readonly): Group the data by the given column in the
metadata. For example, `cluster`.
Defaults to `ScrnaMetabolicLandscape.group_by`
fgsea_args (type=json): Other arguments for the `fgsea::fgsea()` function.
For example, `{"minSize": 15, "maxSize": 500}`.
See <https://rdrr.io/bioc/fgsea/man/fgsea.html> for more details.
plots (type=json): The plots to generate.
Names will be used as the title for the plot. Values will be the arguments
passed to `biopipen.utils::VizGSEA()` function.
See <https://pwwang.github.io/biopipen.utils.R/reference/VizGSEA.html>.
cases (type=json): Multiple cases for the analysis.
If you only have one case, you can specify the parameters directly to
`envs.subset_by`, `envs.group_by`, `envs.fgsea_args`, `envs.plots`,
`envs.select_pcs`, and `envs.pathway_pval_cutoff`.
The name of this default case will be `envs.subset_by`.
If you have multiple cases, you can specify the parameters for each case
in a dictionary. The keys will be the names of the cases and the values
will be dictionaries with the parameters for each case, where the values
will be inherited from `envs.subset_by`, `envs.group_by`, `envs.fgsea_args`,
`envs.plots`, `envs.select_pcs`, and `envs.pathway_pval_cutoff`.
""" # noqa: E501
input = "sobjfile:file"
output = "outdir:dir:{{in.sobjfile | stem}}.pathwayhetero"
lang = config.lang.rscript
envs = {
"gmtfile": None,
"select_pcs": 0.8,
"pathway_pval_cutoff": 0.01,
"ncores": config.misc.ncores,
"subset_by": None,
"group_by": None,
"fgsea_args": {"scoreType": "std", "nproc": 1},
"plots": {
"Pathway Heterogeneity": {
"plot_type": "dot",
"devpars": {"res": 100},
},
},
"cases": {},
}
script = (
"file://../scripts/scrna_metabolic_landscape/"
"MetabolicPathwayHeterogeneity.R"
)
plugin_opts = {
"report": (
"file://../reports/scrna_metabolic_landscape/"
"MetabolicPathwayHeterogeneity.svelte"
)
}
class ScrnaMetabolicLandscape(ProcGroup):DOCS
"""Metabolic landscape analysis for scRNA-seq data
An abstract from
<https://github.com/LocasaleLab/Single-Cell-Metabolic-Landscape>
See docs here for more details
<https://pwwang.github.io/biopipen/pipelines/scrna_metabolic_landscape>
Reference:
Xiao, Zhengtao, Ziwei Dai, and Jason W. Locasale.
"Metabolic landscape of the tumor microenvironment at
single cell resolution." Nature communications 10.1 (2019): 1-12.
Args:
metafile: Either a metafile or an rds file of a Seurat object.
If it is a metafile, it should have two columns: `Sample` and
`RNAData`. `Sample` should be the first column with unique
identifiers for the samples and `RNAData` indicates where the
barcodes, genes, expression matrices are. The data will be loaded
and an unsupervised clustering will be done.
Currently only 10X data is supported.
If it is an rds file, the seurat object will be used directly
is_seurat (flag): Whether the input `metafile` is a seurat object.
If `metafile` is specified directly, this option will be ignored
and will be inferred from the file extension. If `metafile` is
not specified, meaning `<pipeline>.procs.MetabolicInput` is
dependent on other processes, this option will be used to determine
whether the input is a seurat object or not.
noimpute (flag): Whether to do imputation for the dropouts.
If True, the values will be left as is.
gmtfile: The GMT file with the metabolic pathways. The gene names should
match the gene names in the gene list in RNAData or
the Seurat object.
You can also provide a URL to the GMT file.
For example, from
<https://download.baderlab.org/EM_Genesets/current_release/Human/symbol/>.
subset_by (pgarg;type=auto;readonly): Subset the data by the given column in the
metadata. For example, `Response`.
`NA` values will be removed in this column.
If None, the data will not be subsetted.
Multiple columns can be provided as a list, which is helpful when
subsets have overlapping cells.
group_by (pgarg;readonly): Group the data by the given column in the
metadata. For example, `cluster`.
mutaters (type=json): Add new columns to the metadata for
grouping/subsetting.
They are passed to `sobj@meta.data |> mutate(...)`. For example,
`{"timepoint": "if_else(treatment == 'control', 'pre', 'post')"}`
will add a new column `timepoint` to the metadata with values of
`pre` and `post` based on the `treatment` column.
subset (pgarg): Subset the data before analysis.
ncores (type=int): Number of cores to use for parallelization for
each process
"""
DEFAULTS = Diot(
metafile=None,
is_seurat=None,
gmtfile=None,
mutaters=None,
subset=None,
noimpute=True,
ncores=config.misc.ncores,
subset_by=None,
group_by=None,
)
def post_init(self):DOCS
"""Load runtime processes"""
if self.opts.metafile:
suffix = Path(self.opts.metafile).suffix
self.opts.is_seurat = suffix in (".rds", ".RDS", ".qs", ".qs2")
@ProcGroup.add_proc # type: ignore
def p_input(self) -> Type[Proc]:
"""Build MetabolicInputs process"""
from .misc import File2Proc
@mark(board_config_hidden=True)
class MetabolicInput(File2Proc):
"""This process takes Seurat object as input and pass it to the next
processes in the `ScrnaMetabolicLandscape` group.
There is no configuration for this process.
"""
if self.opts.metafile:
input_data = [self.opts.metafile]
return MetabolicInput
@ProcGroup.add_proc # type: ignore
def p_preparing(self) -> Type[Proc] | None:
"""Build SeuratPreparing process"""
if self.opts.is_seurat:
return None
from .scrna import SeuratPreparing
class MetabolicSeuratPreparing(SeuratPreparing):
requires = self.p_input
return MetabolicSeuratPreparing
@ProcGroup.add_proc # type: ignore
def p_clustering(self) -> Type[Proc]:
"""Build SeuratClustering process"""
if self.opts.is_seurat:
return self.p_input # type: ignore
from .scrna import SeuratClustering
class MetabolicSeuratClustering(SeuratClustering):
requires = self.p_preparing
return MetabolicSeuratClustering
@ProcGroup.add_proc # type: ignore
def p_mutater(self) -> Type[Proc]:
"""Build SeuratMetadataMutater process"""
if not self.opts.mutaters and not self.opts.subset:
return self.p_clustering # type: ignore
from .scrna import SeuratMetadataMutater
class MetabolicSeuratMetadataMutater(SeuratMetadataMutater):
requires = self.p_clustering
input_data = lambda ch: tibble(
srtobj=ch.iloc[:, 0],
metafile=[None],
)
envs = {"mutaters": self.opts.mutaters, "subset": self.opts.subset}
return MetabolicSeuratMetadataMutater
@ProcGroup.add_proc # type: ignore
def p_expr_impute(self) -> Type[Proc]:
"""Build process"""
if self.opts.noimpute:
return self.p_mutater # type: ignore
from .scrna import ExprImputation
@annotate.format_doc(indent=3) # type: ignore
class MetabolicExprImputation(ExprImputation):
"""{{Summary}}
You can turn off the imputation by setting the `noimpute` option
of the process group to `True`.
"""
requires = self.p_mutater
return MetabolicExprImputation
@ProcGroup.add_proc # type: ignore
def p_pathway_activity(self) -> Type[Proc]:
"""Build MetabolicPathwayActivity process"""
return Proc.from_proc( # type: ignore
MetabolicPathwayActivity,
"MetabolicPathwayActivity",
requires=self.p_expr_impute, # type: ignore
order=-1,
envs_depth=5,
envs={
"ncores": self.opts.ncores,
"gmtfile": self.opts.gmtfile,
"group_by": self.opts.group_by,
"subset_by": self.opts.subset_by,
},
)
@ProcGroup.add_proc # type: ignore
def p_pathway_heterogeneity(self) -> Type[Proc]:
"""Build MetabolicPathwayHeterogeneity process"""
return Proc.from_proc( # type: ignore
MetabolicPathwayHeterogeneity,
"MetabolicPathwayHeterogeneity",
requires=self.p_mutater, # type: ignore
envs_depth=5,
envs={
"ncores": self.opts.ncores,
"gmtfile": self.opts.gmtfile,
"group_by": self.opts.group_by,
"subset_by": self.opts.subset_by,
},
)
@ProcGroup.add_proc # type: ignore
def p_features(self) -> Type[Proc]:
"""Build MetabolicFeatures process"""
return Proc.from_proc( # type: ignore
MetabolicFeatures,
"MetabolicFeatures",
requires=self.p_expr_impute, # type: ignore
envs_depth=5,
envs={
"ncores": self.opts.ncores,
"gmtfile": self.opts.gmtfile,
"group_by": self.opts.group_by,
"subset_by": self.opts.subset_by,
},
)
if __name__ == "__main__":
ScrnaMetabolicLandscape().as_pipen().run()